- The paper presents PocketVina, a framework that combines machine learning-based pocket prediction with GPU acceleration to achieve state-of-the-art physical docking accuracy, including a 50.96% success rate on PDBbind2020.
- The method integrates P2Rank and QuickVina 2-GPU 2.1 to efficiently predict and validate ligand-binding poses, demonstrating robust generalization to novel protein-ligand complexes.
- The results highlight PocketVina's effectiveness in handling ligand flexibility and scalability, positioning it as a vital tool for high-throughput virtual screening in drug discovery.
PocketVina: Advancements in Molecular Docking
Introduction
Molecular docking serves as an essential computational approach in drug discovery, primarily involved in predicting the interaction of small molecules with target proteins. The paper, "PocketVina Enables Scalable and Highly Accurate Physically Valid Docking through Multi-Pocket Conditioning" (2506.20043), introduces PocketVina, a novel docking framework that integrates pocket prediction with systematic exploration. It aims to enhance the prediction of ligand-binding poses while maintaining high accuracy and scalability.
Methodology
PocketVina integrates two main components: P2Rank for protein pocket prediction and QuickVina 2-GPU 2.1 for molecular docking. P2Rank utilizes a machine learning model to predict ligandable pockets on a protein's solvent-accessible surface, identifying and classifying pockets based on ligandability scores. QuickVina 2-GPU 2.1, an optimized extension of AutoDock Vina, employs GPU acceleration to perform docking at predicted pocket centers with increased efficiency. This dual-step process allows PocketVina to rapidly generate and validate ligand conformations.
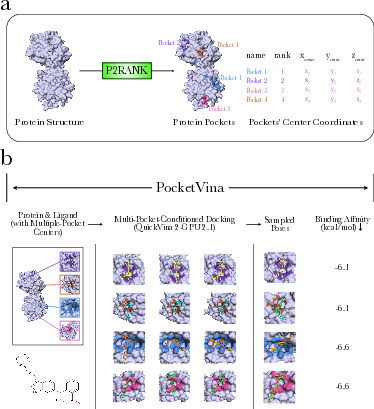
Figure 1: Overview of PocketVina showcasing the multi-pocket conditioning process and the synergy of P2Rank and QuickVina 2-GPU 2.1.
Results
PocketVina showcased superior performance across several benchmarks including PDBbind2020, Astex, PoseBusters, and DockGen datasets. It achieved a notable state-of-the-art success rate when evaluated for physically valid docking poses, outperforming traditional and contemporary deep learning-based methods. In the PDBbind2020 test set, PocketVina achieved a success rate of 50.96% under stringent criteria combining ligand r.m.s.d. measurements and physical validity benchmarks.
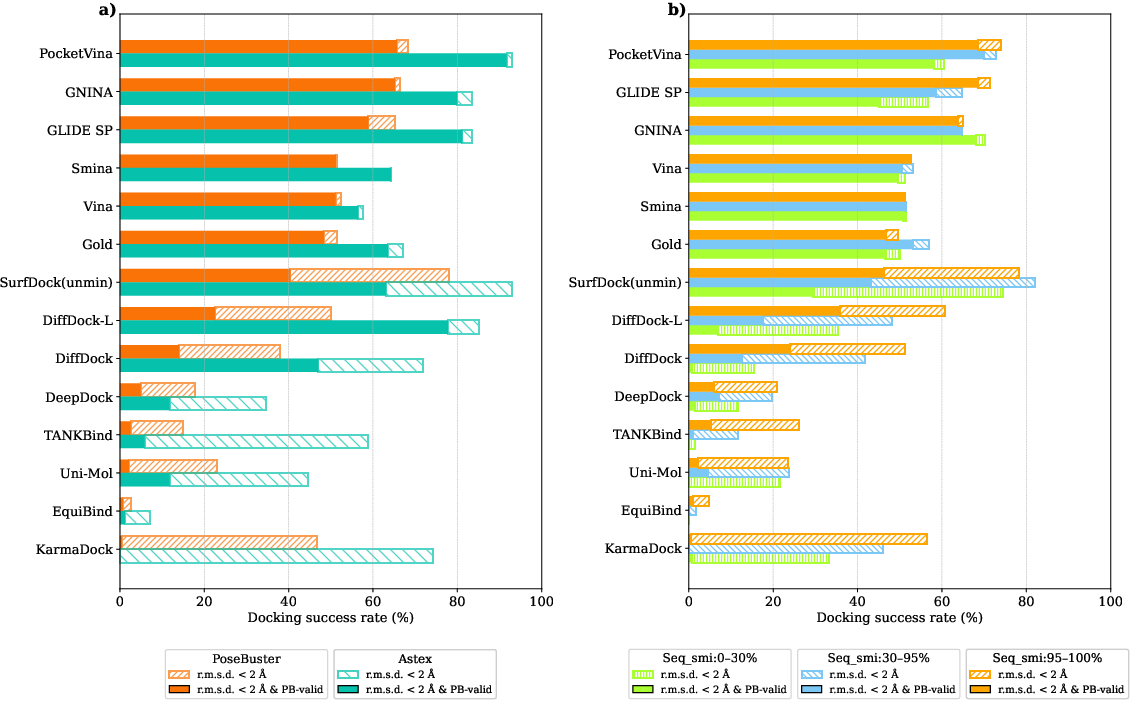
Figure 2: Comparative performance of PocketVina in terms of success rates across different datasets, indicating its effectiveness in generating physically valid docking poses.
Generalization Capabilities
Notably, PocketVina demonstrated robust generalization capabilities, effectively processing structurally diverse and previously unseen protein-ligand complexes. The method maintained superior docking accuracy even with novel proteins outside of typical training datasets, thus underscoring its utility in drug discovery scenarios where novel target interactions are paramount.
Impact of Ligand Flexibility
An essential aspect of molecular docking is handling ligand flexibility. PocketVina exhibited consistent high performance across ligands of varying flexibility. For rigid ligands (≤5 rotatable bonds), PocketVina achieved nearly 80% success in delivering physically valid poses. This ability to efficiently handle flexible ligands further advocates for its deployment in high-throughput virtual screening, bolstering the structure-based drug design workflows.
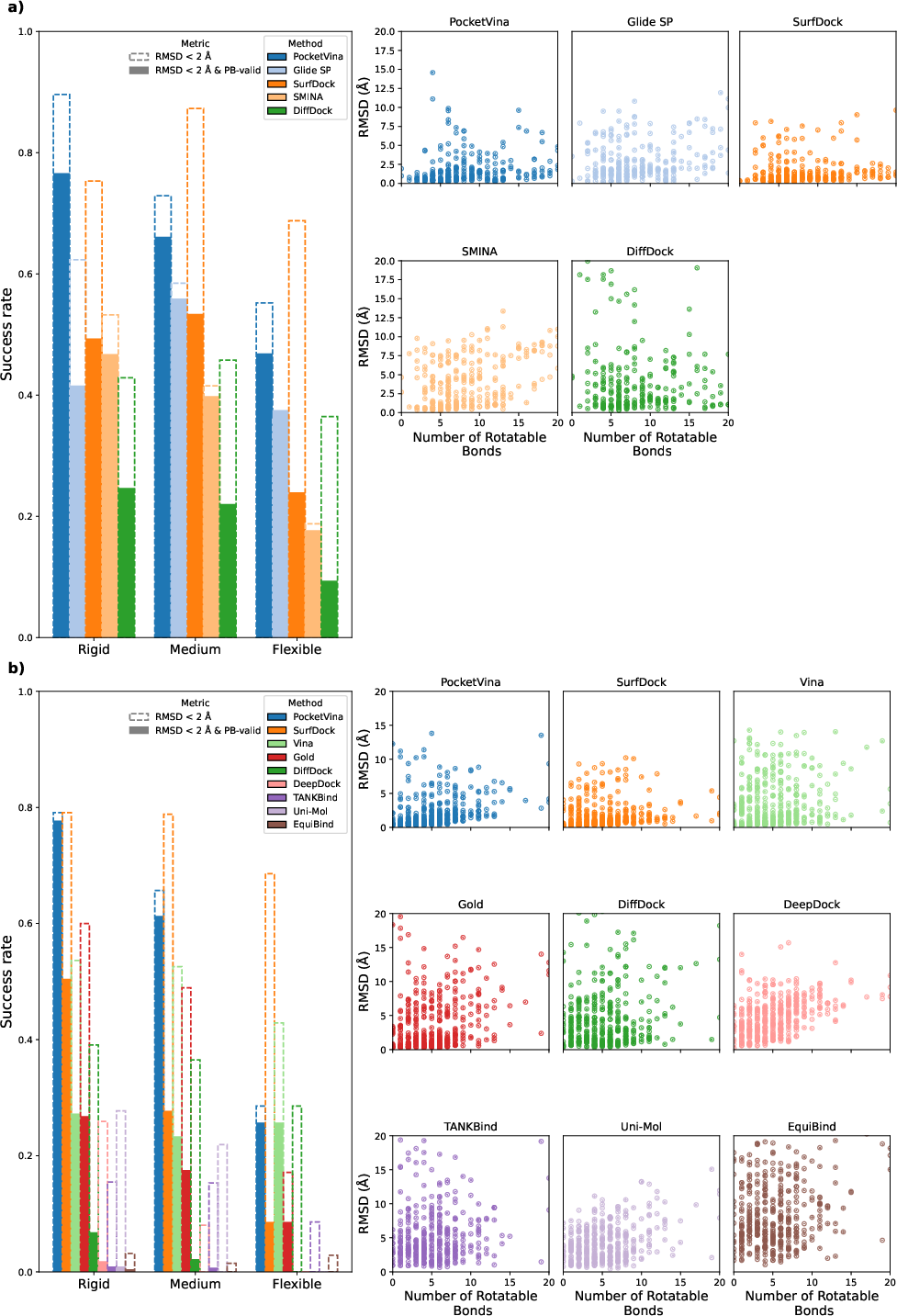
Figure 3: Analysis of docking success rates across ligand flexibility categories, illustrating PocketVina's efficacy.
Future Directions and Conclusion
PocketVina's introduction marks a significant advancement in the computational docking field by elegantly balancing computational efficiency with high docking accuracy. Its integration of pocket-centred docking targets a critical limitation in traditional approaches, and the framework's capability to process large-scale protein-ligand datasets rapidly positions it as a favorable tool for early drug discovery phases.
In conclusion, PocketVina not only exemplifies methodological innovation through integrating machine learning with classical docking approaches but also establishes a scalable solution suitable for modern drug discovery pipelines. Future work could focus on further enhancing the framework's adaptability to even broader chemical spaces and deeper integration with AI-driven compound optimization strategies.
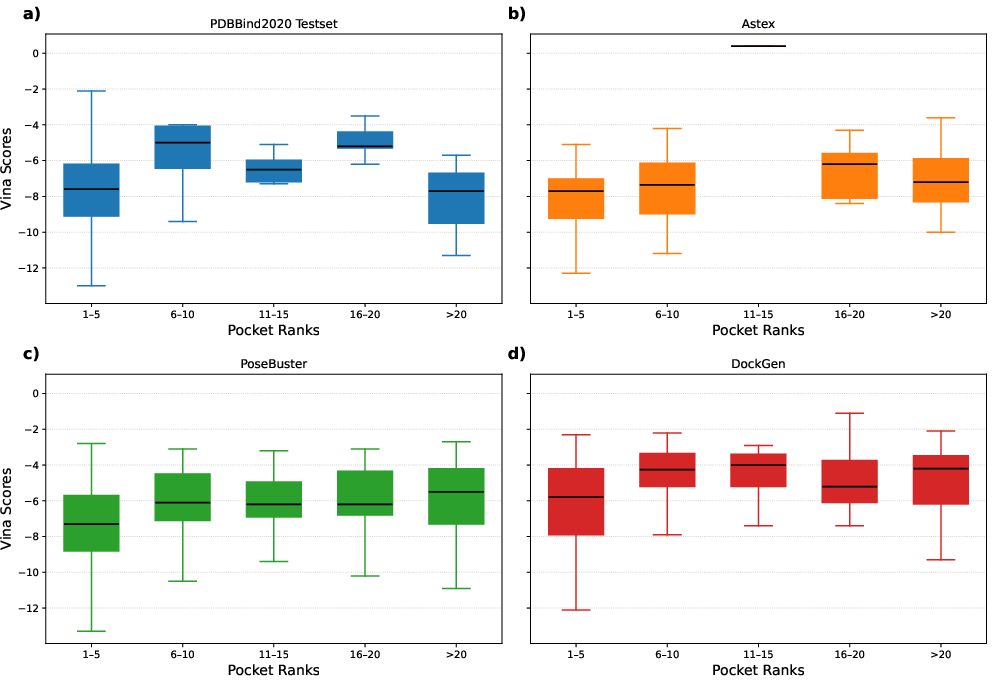
Figure 4: Visualization of the relationship between pocket rankings and predicted binding affinities, affirming PocketVina's efficient resource allocation in docking tasks.

