- The paper introduces CANet, a unified framework integrating commutative algebra embeddings with deep learning to predict mutation impacts on disease and protein properties.
- It demonstrates state-of-the-art performance with notable improvements in MCC, AUC, PCC, and normalized accuracies across disease, stability, and solubility tasks.
- The framework provides mechanistic insights by linking algebraic invariants to physical interactions like hydrogen bonds and salt bridges, enhancing interpretability through XAI.
Commutative Algebra Neural Network for Elucidating Genetic Disease Mechanisms
Introduction and Motivation
The paper introduces a unified, interpretable machine learning framework—Commutative Algebra Neural Network (CANet)—for predicting the effects of genetic mutations on protein function, specifically targeting disease association, protein stability, and solubility changes. The approach leverages multiscale commutative algebra, particularly Persistent Stanley–Reisner Theory (PSRT), to extract algebraic invariants from protein structures, integrating these with transformer-based sequence embeddings and auxiliary physical features. This framework addresses the limitations of prior models, which often lack interpretability, fail to integrate physical/chemical interactions, and treat the three prediction tasks in isolation.
CANet Workflow and Algebraic Embedding
CANet's workflow begins with 3D protein structures, from which both wild-type and mutant forms are generated. Around each mutation site, element-specific atom subsets are extracted to form local subcomplexes. Multiscale commutative algebra embeddings are computed by tracking the evolution of facet ideals and f-vector curves under a filtration process, capturing both geometric and chemical perturbations induced by mutations. These algebraic features are concatenated with auxiliary descriptors (e.g., solvent-accessible surface area, secondary structure) and ESM-2 transformer embeddings to form the input for downstream models: a deep neural network (CANet) and a gradient boosting tree (CATree).
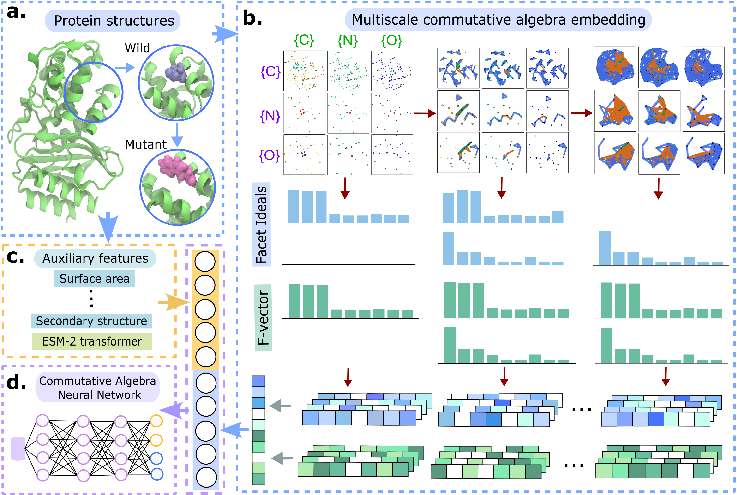
Figure 1: CANet workflow, from 3D structure and mutant generation to multiscale commutative algebra embedding and feature integration for downstream prediction.
The algebraic embedding is constructed using element- and site-specific atom sets, with a modified Euclidean distance to focus on interactions between mutation sites and their neighborhoods. The PSRT framework generates a filtration of simplicial complexes, from which persistent facet ideals and f-vectors are extracted, providing a rigorous, interpretable representation of local and global structural changes.
Disease-Associated Mutation Prediction
On the M546 dataset (transmembrane protein mutations), CATree achieves a blind test MCC of 0.86, a 7.5% improvement over the persistent homology-based TopGBT model, and an AUC of 0.96. In 10-fold cross-validation, CATree attains an MCC of 0.78 and an F1-score of 0.92, outperforming all existing state-of-the-art models. These results demonstrate robust generalization, particularly in settings where previous models exhibited overfitting.
Protein Stability Change Prediction
For mutation-induced stability changes, CANet is evaluated on the S2648 and S350 datasets. On S2648, CANet achieves a PCC of 0.82 (6.49% higher than TNet-MP-2) and an RMSE of 0.85 (9.6% improvement). On the S350 benchmark, CANet maintains a PCC of 0.82, outperforming TNet-MP-2 by 1.23%. CATree also demonstrates competitive performance, especially on smaller datasets.
Protein Solubility Change Classification
On the PON-Sol2 dataset, CANet and CATree achieve normalized accuracies of 0.702 and 0.700, respectively, representing up to 7.01% improvement over PON-Sol2 models and 2.93% over TopGBT. In blind test classification, CANet achieves a normalized accuracy of 0.580, up to 6.4% higher than PON-Sol2. The models also exhibit superior GC2 scores, indicating robustness in class-imbalanced settings.
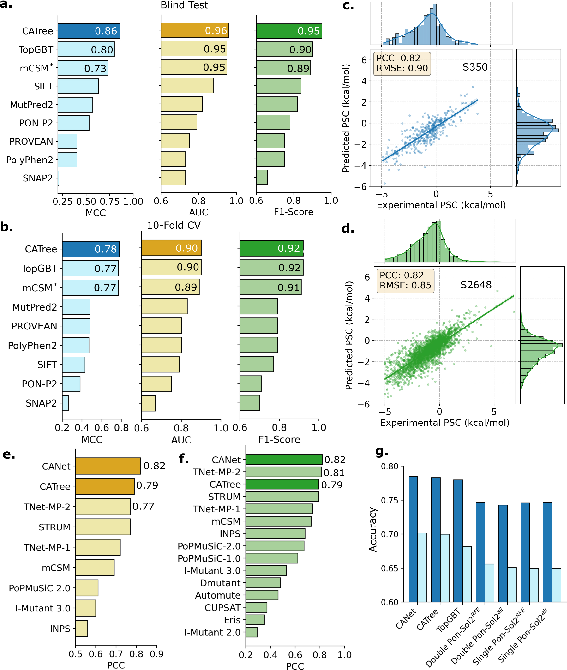
Figure 2: Comparative performance of CANet and CATree across disease, stability, and solubility prediction tasks, highlighting improvements over prior state-of-the-art models.
Interpretability and Mechanistic Insights
A central contribution of the framework is its interpretability. The commutative algebraic features—persistent facet ideals and f-vectors—can be directly mapped to physical and chemical interactions, such as hydrogen bonds, salt bridges, and electrostatic shifts. This enables mechanistic tracing of mutation effects, supporting eXplainable AI (XAI) in molecular biophysics.
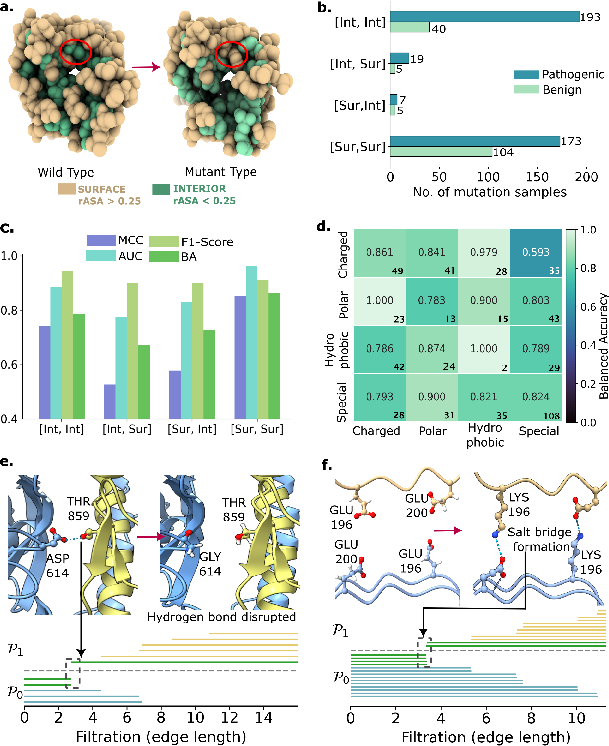
Figure 3: Electrostatic interaction analysis and mutation impact on protein structure and pathogenicity, with persistent facet ideals revealing hydrogen bond and salt bridge formation/disruption.
The analysis demonstrates that CANet can identify the loss of hydrogen bonds (e.g., D614G in SARS-CoV-2 spike protein) and the formation of new salt bridges (e.g., E196K in prion protein), both of which are critical for understanding disease mechanisms. The algebraic invariants provide a transparent link between model predictions and underlying molecular events.
Structural Motif Analysis and Scalability
The paper further illustrates the interpretability of commutative algebraic features by analyzing canonical structural motifs (alpha-helices, beta-sheets, DNA) using Rips complex-based filtrations. Persistent facet barcodes and f-vector curves capture the emergence and disappearance of geometric features at different scales, reflecting the underlying biophysical organization.
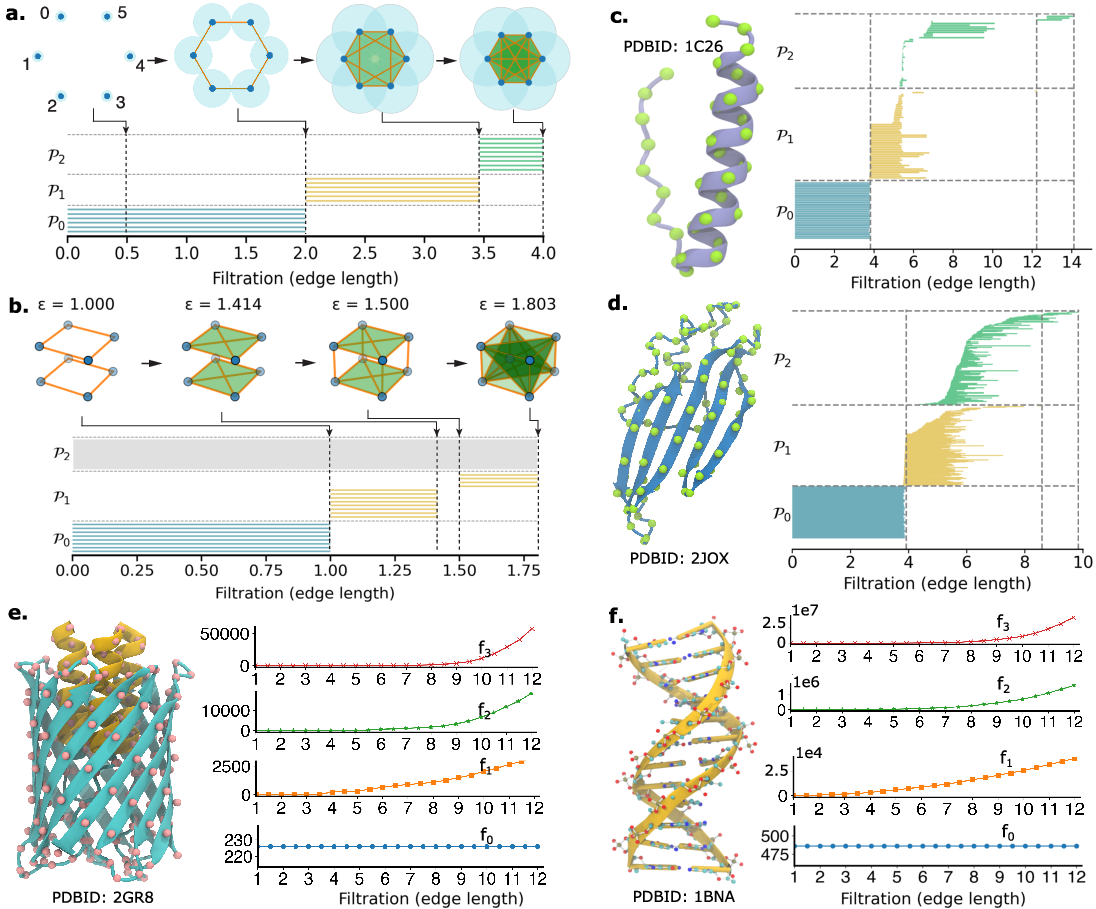
Figure 4: Multiscale commutative algebra analysis on point-cloud data, with facet persistence barcodes and f-vector curves for simple geometric configurations and biomolecular structures.
For large biomolecular systems, f-vector analysis provides a scalable alternative to full barcode computation, enabling the application of CANet to high-dimensional structural data.
Biological and Biochemical Context Dependence
The study stratifies predictive performance by mutation region (surface vs. interior) and mutation type (charged, polar, hydrophobic, special). CATree achieves higher balanced accuracy for [Int, Int] and [Sur, Sur] mutations, with lower performance in mixed-region categories, likely due to limited sample sizes. The model is particularly sensitive to mutations that induce significant electrostatic or hydrophobic changes, consistent with known pathogenic mechanisms.
Implications and Future Directions
The integration of commutative algebra with deep learning and transformer-based embeddings establishes a new paradigm for interpretable, mechanistically grounded prediction of mutation effects. The algebraic framework provides a rigorous mathematical foundation for XAI in molecular biology, enabling the tracing of predictions to specific structural and chemical perturbations.
Potential future developments include:
- Extension to co-evolutionary and allosteric effects, leveraging the algebraic invariants to capture long-range structural dependencies.
- Application to other molecular systems (e.g., nucleic acids, protein–ligand complexes) where local and global geometric features are critical.
- Further integration with topological data analysis (TDA) and spectral methods to enhance sensitivity to both local and global structural changes.
- Development of scalable algorithms for large-scale biomolecular datasets, leveraging f-vector and other summary statistics.
Conclusion
The commutative algebra neural network framework provides a unified, interpretable, and generalizable approach for predicting the effects of genetic mutations on protein function. By embedding multiscale algebraic invariants into machine learning models, the approach achieves state-of-the-art performance across disease, stability, and solubility prediction tasks, while offering mechanistic insights into the molecular basis of genetic diseases. The algebraic perspective opens new avenues for XAI in computational biology and has broad implications for the development of interpretable AI in the life sciences.