- The paper presents a unified generative paradigm that integrates diffusion-pretrained structure encoding with autoregressive sequence generation to enhance structure-based drug design.
- It applies Direct Preference Optimization and VAE-based noise injection to optimize binding affinity, drug-likeness, and diversity of generated molecules.
- Experimental results on CrossDocked2020 benchmarks show that MolChord outperforms baselines in generating chemically plausible, high-affinity ligands with efficient inference.
MolChord: Structure-Sequence Alignment for Protein-Guided Drug Design
Introduction and Motivation
Structure-based drug design (SBDD) requires precise alignment between protein structural representations and molecular ligand representations, with the additional constraint that generated compounds must exhibit favorable pharmacological properties. The MolChord framework addresses two persistent challenges in SBDD: (1) the limited availability of high-quality protein–ligand pairs for supervised learning, and (2) the difficulty of aligning generated molecules with desired drug-like properties. MolChord leverages a unified generative paradigm, integrating a diffusion-based structure encoder and an autoregressive sequence generator, and refines alignment through Direct Preference Optimization (DPO) on a property-aware dataset.
Model Architecture
MolChord comprises three principal modules: a diffusion-pretrained structure encoder, an autoregressive sequence generator, and a lightweight adapter with an auxiliary VAE for alignment and diversity enhancement.
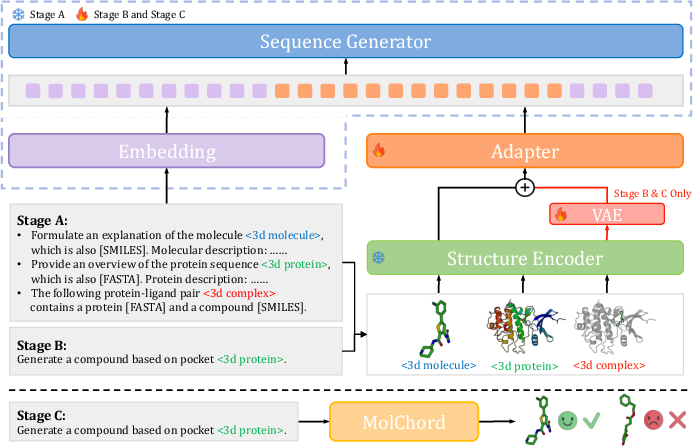
Figure 1: Overview of MolChord, illustrating the integration of structure encoder, sequence generator, VAE-based perturbation, and DPO-based preference optimization.
Structure Encoder
The encoder is based on the FlexRibbon framework, utilizing a Transformer variant with geometric inductive biases. It processes proteins at the residue level, molecules at the atom level, and complexes as joint residue–atom graphs. Pretraining is performed on large-scale protein and molecule datasets (AlphaFoldDB, PDB), with diffusion objectives to capture spatial and sequential context.
Sequence Generator
The generator is a NatureLM variant, pretrained on protein FASTA, molecular SMILES, and textual annotations. It employs a next-token prediction objective, supporting cross-modal generation and alignment. The tokenizer is extended to handle domain-specific tokens for proteins and molecules.
Adapter and VAE
The adapter is a gated MLP that projects encoder outputs into the generator’s embedding space. The VAE introduces stochasticity during supervised fine-tuning, perturbing protein features to enhance diversity and robustness in ligand generation.
Structure-Sequence Alignment
Inputs are interleaved sequences of text, structure tokens, and annotations. Structural entities (e.g., ⟨3dprotein⟩) are processed by the encoder and injected into the generator’s input embedding, enabling joint attention over structural and symbolic features.
Training Strategy
MolChord employs a three-stage training protocol:
- Stage A: Adapter alignment via next-token prediction on large-scale protein, molecule, and complex datasets. Encoder and generator weights are frozen.
- Stage B: Supervised fine-tuning on protein–ligand complexes, with VAE-based noise injection to promote diversity.
- Stage C: DPO-based reinforcement learning on a curated subset of CrossDocked2020, optimizing for binding affinity, drug-likeness, and synthesizability.
Data partitioning ensures that SFT and DPO operate on disjoint sets, with diversity-based filtering for preference optimization.
Experimental Results
MolChord is evaluated on the CrossDocked2020 benchmark, compared against diffusion, flow, and autoregressive baselines. Metrics include Vina Dock (binding affinity), QED, SA, diversity, and success rate (joint drug-like, synthesizable, high-affinity criteria).
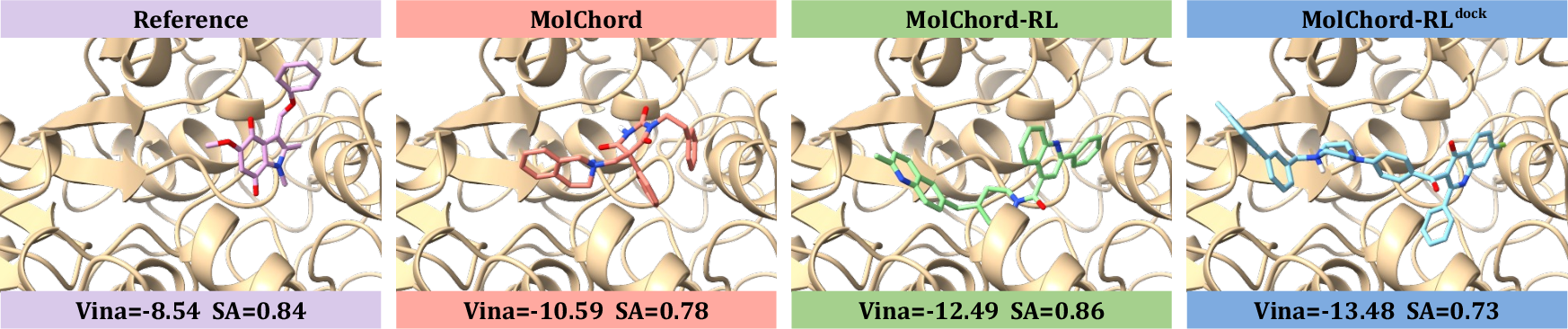
Figure 2: Visualizations of reference molecules and ligands generated by MolChord, MolChord-RL, and MolChord-RLdock, highlighting improvements in affinity and molecular properties.
MolChord and its RL variants achieve state-of-the-art performance across all key metrics. Notably, MolChord-RL surpasses the 70% high-affinity threshold, outperforming strong baselines such as FlowSBDD and DecompDiff. QED and SA scores are maximized without sacrificing diversity, and the success rate is substantially higher than prior methods.
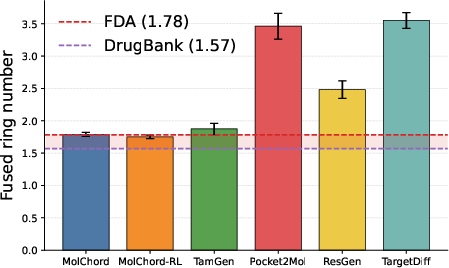
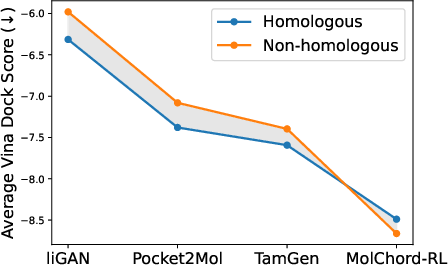
Figure 3: Barplot of fused ring counts in top-ranked compounds, demonstrating MolChord’s regularization towards chemically plausible ring systems.
MolChord matches the fused ring distribution of FDA-approved drugs, avoiding the overproduction of complex ring systems observed in other models. This regularization is critical for synthetic accessibility and toxicity reduction.
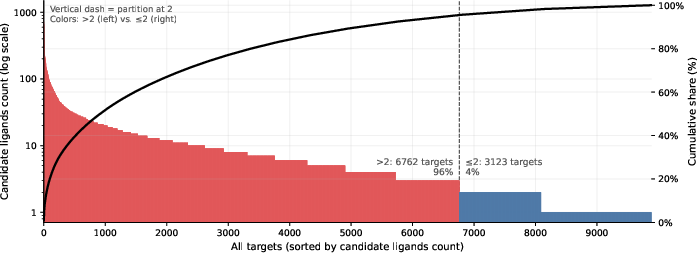
Figure 4: Distribution of candidate ligands per target in CrossDocked2020, illustrating the stratified data partitioning for SFT and DPO.
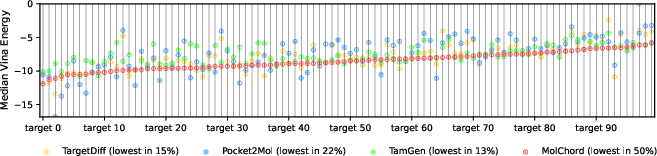
Figure 5: Median Vina energy comparison across models, with MolChord generating the highest-affinity molecules for the majority of targets.
Out-of-Distribution Generalization
MolChord-RL demonstrates robust generalization to non-homologous proteins, with improved affinity scores compared to homologous cases. This is attributed to the structure encoder’s large-scale pretraining and cross-modal alignment, enabling transferability beyond the training distribution.
Ablation Studies
Ablations confirm the necessity of full structure-sequence alignment and diversity-based data partitioning. Removal of the diffusion-pretrained encoder or DPO fine-tuning degrades the affinity–drug-likeness trade-off. Incorporation of the VAE consistently improves diversity and affinity metrics.
Implementation Considerations
- Computational Requirements: Pretraining the structure encoder requires substantial resources (128 A100 GPUs, two weeks). Adapter alignment and SFT are less demanding, while DPO is highly efficient.
- Inference Efficiency: MolChord generates 100 compounds per target in ~4 seconds on a single A100 GPU, outperforming previous approaches in throughput.
- Deployment: The modular design allows for flexible integration into SBDD pipelines, with the adapter enabling rapid adaptation to new protein or molecule modalities.
Implications and Future Directions
MolChord establishes a unified framework for SBDD, integrating geometric and sequential modalities with principled preference optimization. The approach demonstrates that large-scale cross-modal pretraining, combined with lightweight alignment and RL-based fine-tuning, yields models that are both accurate and chemically plausible. The robust generalization to out-of-distribution targets and efficient inference suggest practical utility in real-world drug discovery workflows.
Future work may explore:
- Extension to multi-target or polypharmacology scenarios.
- Incorporation of additional modalities (e.g., RNA, materials) via further expansion of the generative backbone.
- Integration with experimental feedback loops for closed-cycle optimization.
- Scaling to larger model capacities and more diverse chemical spaces.
Conclusion
MolChord advances the state-of-the-art in protein-guided drug design by unifying diffusion-based structure encoding and autoregressive sequence generation, with explicit alignment and preference optimization. The framework achieves strong performance across binding affinity, drug-likeness, synthesizability, and diversity, and demonstrates robust generalization and efficiency. These results underscore the value of cross-modal alignment and RL-based optimization in SBDD, providing a scalable and practical foundation for future AI-driven drug discovery.

