- The paper combined MD simulations with experimental techniques to accurately extract conductivity, transference number, thermodynamic factor, and salt diffusion coefficients in carbonate-based electrolytes.
- The hybrid methodology leverages Onsager transport theory and reference frame alignment to reconcile discrepancies in traditional Li metal interface measurements.
- The study provides a scalable framework for benchmarking transport parameters, aiding battery model calibration and the rational design of new electrolyte formulations.
Integrative Parametrization of Carbonate-Based Electrolytes: MD and Experimental Synthesis
Introduction
The study, "Combining Molecular Dynamics and Experimental Methods for the Parametrization of Binary Carbonate-Based Electrolytes" (2512.13225), presents a systematic framework for determining the fundamental transport and thermodynamic parameters of nonaqueous carbonate-based battery electrolytes. The approach strategically combines molecular dynamics (MD) simulations with electrochemical measurements and electrophoretic NMR (eNMR), explicitly targeting limitations in traditional experimental protocols caused by interphasial effects at Li metal interfaces. The paper’s method enables the accurate decomposition and assignment of the conductivity (κ), transference number (t+), thermodynamic factor (TDF), and salt diffusion coefficient (D±) for LiPF6 in systems of varying carbonate solvent compositions over a wide concentration and temperature range.
Methodology: Hybrid MD-Experimental Protocol
The protocol’s foundation lies in Onsager transport theory. The authors leverage the Fong et al. framework for MD-based determination of Onsager coefficients via Green-Kubo relations, enabling extraction of κ, t+, and D± directly from time-correlated fluxes. Importantly, parameters are computed in the volume (VOL) reference frame for consistency with eNMR and concentration cell outputs, mitigating discrepancies stemming from choice of reference (e.g., center-of-mass).
Experimental data—electrochemical impedance spectroscopy (EIS) for conductivity and concentration cell measurements for the mixed parameter a(c,T)=(1−t+)TDF—are then combined with the MD-obtained t+ for explicit TDF deconvolution. Independent validation of t+ is provided via eNMR, offering a non-interphasial measurement directly in the bulk.
The study examines three technically relevant electrolytes:
- EC:EMC (3:7, weight)
- EC:DMC:PC (27:63:10, volume)
- EC:EMC:MP (2:6:2, volume)
at −20∘C≤T≤20∘C and 0.1M≤c≤3.0M, capturing both practical and limiting conditions.
Transport Parameter Trends and Reference Frame Alignment
The MD-experimental synergy enables precise benchmarking of computed parameters against literature and new measurements. Key transport parameters exhibit the following characteristics:
- Conductivity (κ): Simulated and measured values concur for c<2M, with all systems exhibiting a pronounced maximum in κ at intermediate salt loadings and monotonic enhancement with temperature, reflecting the interplay of ion association and solvent viscosity reduction.
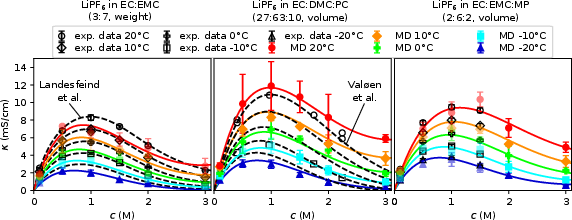
Figure 1: Conductivities κ from MD and experiment as a function of c and T indicate a peak at intermediate salt concentrations.
- Transference Number (t+VOL): Both MD and eNMR show t+VOL values ≈0.36−0.44 across all systems, with only marginal temperature/concentration dependence. This finding is in strong contrast to some literature values obtained via classical Li|electrolyte|Li polarization, which can present artificial negative transference numbers due to interfacial artifacts.
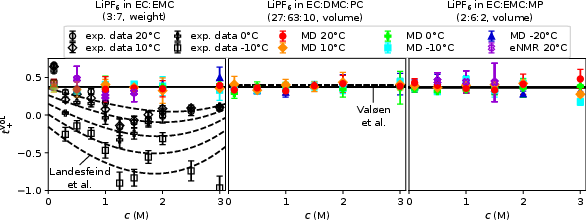
Figure 2: t+VOL from MD and eNMR nearly constant, supporting the dominance of PF6− mobility and invalidating negative t+ reports.
- Thermodynamic Factor (TDF): The extracted TDF rises with c and falls with T, but the scaling matches between systems after accounting for the literature’s inconsistent reference-frame conventions (factor of 2 applied where needed for comparison).
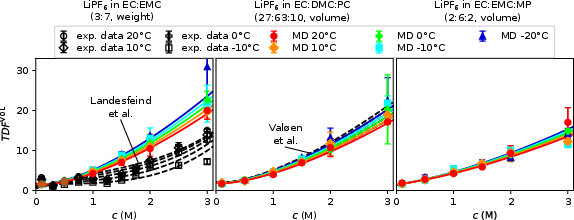
Figure 3: TDF displays a monotonic increase with salt content, in line with increased ion–solvent interactions.
- Salt Diffusion Coefficient (D±VOL): MD predicts complex, non-monotonic D± versus c, matching predictions from advanced electrolyte models but deviating from experimental polarization data—again highlighting the unreliability of Li metal-based measurements in carbonate electrolytes.
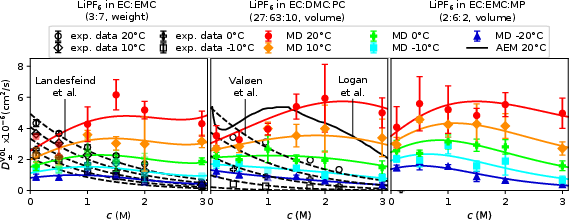
Figure 4: D±VOL from MD reveals intricate dependence on both concentration and temperature.
Analysis of Viscosity, Ion Association, and Diffusion Mechanism
The authors deepen the analysis via MD-quantified viscosity (via Stokes-Einstein), ion association statistics, and solvent-shell exchange metrics. Viscosity is highest in EC:EMC and lowest in EC:EMC:MP, but all systems exhibit expected Arrhenius scaling with temperature and a rise with salt content.
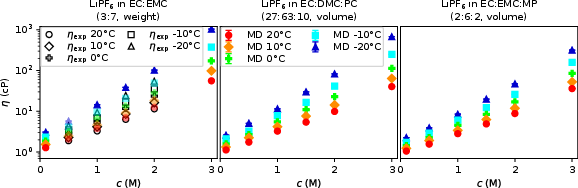
Figure 5: MD-calculated viscosity, confirmed against experiment, reveals strong compositional dependence among binary and ternary solvents.
Analysis of solvent-exchange lengths and the free ion fraction demonstrates:
- Ion transport in these systems is dominated by vehicular mechanisms for Li+ (longer solvent-travel distances in the shell),
- The decrease in free ion content with c and T,
- Only minor formation of higher-order ion aggregates (PTI/NTI), even at high concentration, supporting classic vehicular transport scenarios for both cation and anion.
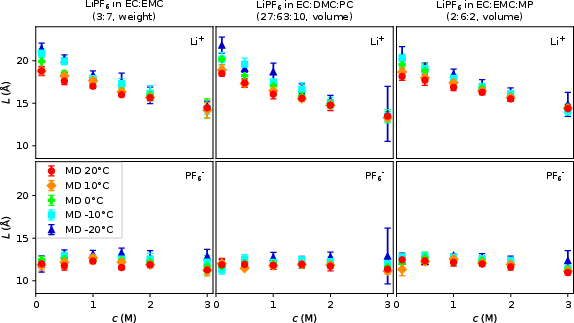
Figure 6: Solvent exchange lengths (diffusion length, L) clarify the relative stability of the ion solvation shells.
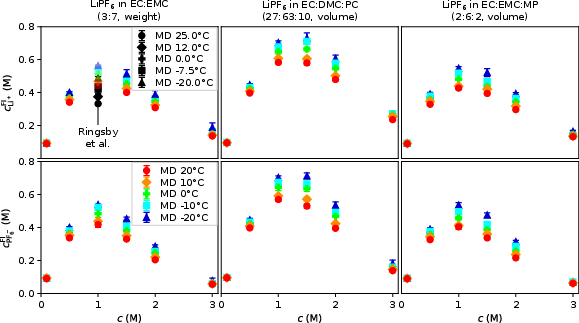
Figure 7: Free ion fraction for Li+ and PF6− as a function of c and T—MI data matches well with literature at moderate concentrations.
Implications and Outlook
This work robustly demonstrates that combining MD with reference-aligned experimental measurements overcomes long-standing challenges in accurately decoding the real transport properties of carbonate-based electrolytes. The separation of interfacial/phase artifacts from bulk electrolyte phenomena is particularly significant for both theoretical calibration of battery models and the rational engineering of future electrolyte systems.
The study's methodology can be further generalized to other multicomponent electrolytes, including emerging systems such as solvate-enhanced or ionic-liquid–blended solutions, provided that robust reference-frame transformations and consistent scaling corrections are maintained. Additionally, the approach holds promise for accelerating the design and screening of new electrolyte formulations using physically calibrated MD-driven property maps, especially as force fields continue to improve with polarization and quantum corrections.
From a modeling standpoint, accurate, artifact-free transport data such as supplied by this study are essential for first-principles battery simulation at both the cell and device level—supporting predictive, physics-based optimization and lifetime modeling.
Conclusion
The hybrid MD-experimental technique systematically developed and validated in this work provides a scalable pathway for electrolyte characterization, circumventing pitfalls inherent to methods dependent on Li metal interfaces. The established methodology yields consistent, reliable transport and thermodynamic parameters for a range of LiPF6 carbonate systems over wide operating conditions. The findings have direct application in battery model calibration and future electrolyte optimization, highlighting the necessity of careful experimental design and comprehensive theoretical modeling for electrolyte science advancement.