A Unification of Discrete, Gaussian, and Simplicial Diffusion
Abstract: To model discrete sequences such as DNA, proteins, and language using diffusion, practitioners must choose between three major methods: diffusion in discrete space, Gaussian diffusion in Euclidean space, or diffusion on the simplex. Despite their shared goal, these models have disparate algorithms, theoretical structures, and tradeoffs: discrete diffusion has the most natural domain, Gaussian diffusion has more mature algorithms, and diffusion on the simplex in principle combines the strengths of the other two but in practice suffers from a numerically unstable stochastic processes. Ideally we could see each of these models as instances of the same underlying framework, and enable practitioners to switch between models for downstream applications. However previous theories have only considered connections in special cases. Here we build a theory unifying all three methods of discrete diffusion as different parameterizations of the same underlying process: the Wright-Fisher population genetics model. In particular, we find simplicial and Gaussian diffusion as two large-population limits. Our theory formally connects the likelihoods and hyperparameters of these models and leverages decades of mathematical genetics literature to unlock stable simplicial diffusion. Finally, we relieve the practitioner of balancing model trade-offs by demonstrating it is possible to train a single model that can perform diffusion in any of these three domains at test time. Our experiments show that Wright-Fisher simplicial diffusion is more stable and outperforms previous simplicial diffusion models on conditional DNA generation. We also show that we can train models on multiple domains at once that are competitive with models trained on any individual domain.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What is this paper about?
This paper brings together three different ways to use “diffusion models” to generate and edit sequences like DNA, proteins, and words. These three ways—discrete diffusion, Gaussian diffusion, and simplicial diffusion—have each had their own pros and cons, but they haven’t been clearly connected. The authors show that all three are actually different views of the same underlying process from population genetics, called the Wright–Fisher model. Using this connection, they fix stability problems, make results comparable, and even train one model that can work across all three settings.
What questions are the authors trying to answer?
The paper focuses on four simple but important questions:
- How are discrete, Gaussian, and simplicial diffusion related in a clear, mathematical way?
- If different models give different kinds of “likelihoods” (a score for how well the model explains data), can we compare them fairly?
- The models use very different “hyperparameters” (settings you choose before training). Can we understand how these correspond across models?
- Simplicial diffusion should be powerful but is notoriously unstable. Can we make it stable and fast?
- Can we train one model that works in all three domains (discrete, Gaussian, and simplicial) at test time, so users don’t have to pick a single approach upfront?
How did they approach the problem?
Think of diffusion modeling like this: you start with a clean sequence (say, a DNA string), add noise step by step (the “forward” process), then train a model to remove the noise (the “reverse” process) so you can generate new realistic sequences or edit existing ones.
The three approaches are:
- Discrete diffusion: you change letters directly (e.g., A→C→T), staying in the original symbol space.
- Gaussian diffusion: you map each letter to a point in continuous space (like coordinates) and add continuous noise.
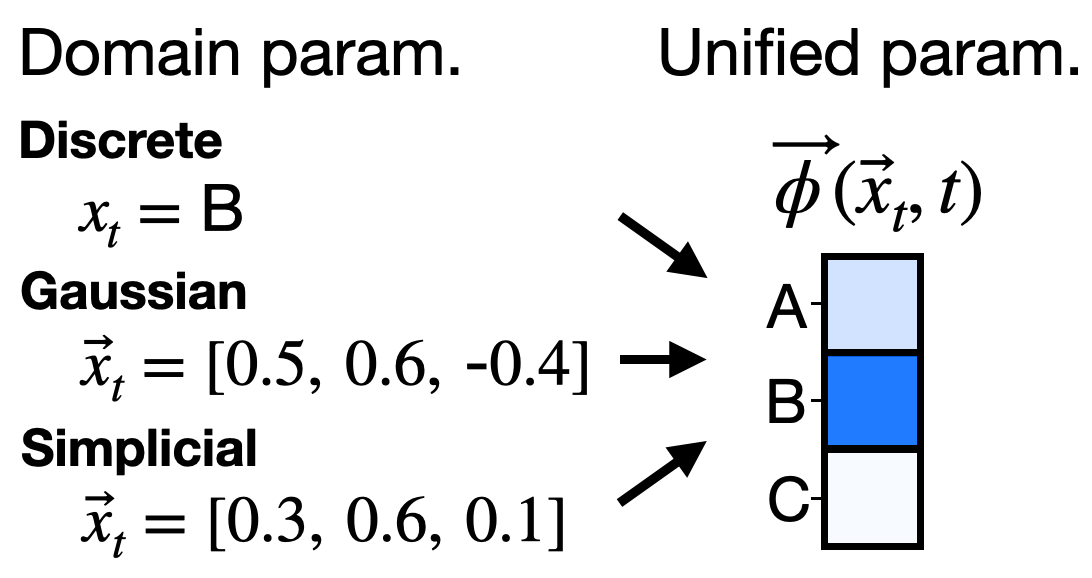
- Simplicial diffusion: you model the probabilities of each letter at a position; these probabilities live on a geometric shape called a simplex (like a triangle for three letters, or a tetrahedron for four).
The authors unify these by using the Wright–Fisher model from genetics:
- Imagine each position in your sequence is a population of individuals, each holding a letter (A, C, G, or T for DNA). Over time, individuals mutate (change letters), and populations reproduce (new generations form from existing ones).
- If the population size is 1, you get discrete diffusion (one letter per position).
- If the population size is huge and there’s no reproduction, the noise looks Gaussian (continuous).
- If the population is huge and there is reproduction, the population’s letter frequencies move across the simplex—that’s simplicial diffusion.
They also use ideas like the central limit theorem (large populations behave like smooth curves), and careful time-scaling (“time dilation”) to align the math across models.
What did they find, and why is it important?
- A single framework: They prove that discrete, Gaussian, and simplicial diffusion are all different parameterizations of Wright–Fisher dynamics. Discrete = population size 1. Gaussian and simplicial = large population limits (without vs. with reproduction).
- Comparable likelihoods: Normally, continuous models (Gaussian) can have infinite training losses near the start of time because the reverse path should be very “deterministic” early on, but the neural net isn’t. The authors fix this with a “hollow parameterization,” which weights the model’s prediction by how much the observed noisy data supports each possible clean input. This removes the singularity and makes likelihoods directly comparable across models.
- Matching hyperparameters: They show how the mutation-rate matrix (for discrete diffusion) determines the right continuous embedding for Gaussian diffusion. In simple terms: the most important directions in the mutation dynamics become the axes in the continuous space. This lets you translate design choices across models and check them visually.
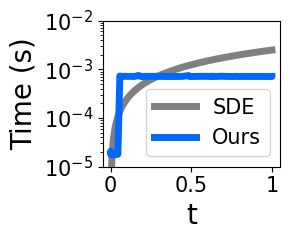
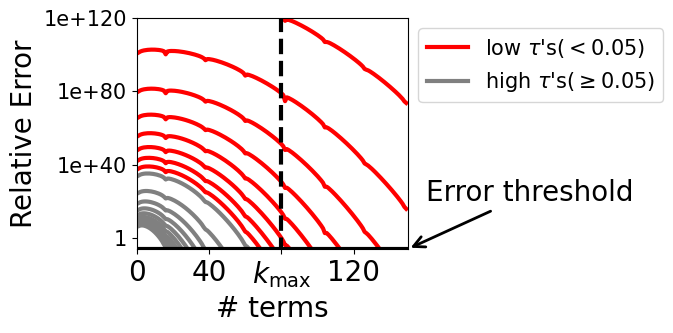
- Stable, fast simplicial diffusion: Simplicial diffusion had numerical issues when time is very small (close to the clean starting point). Using decades of population genetics results, they replace costly, unstable SDE simulation with exact, efficient sampling (via Jenkins’ method) and swap a tricky infinite series for a stable approximation at small times. This makes training practical and robust.
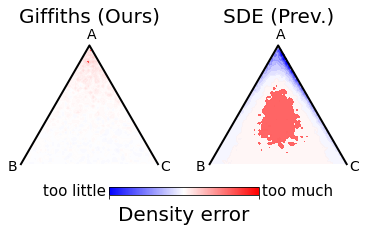
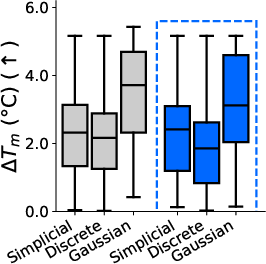
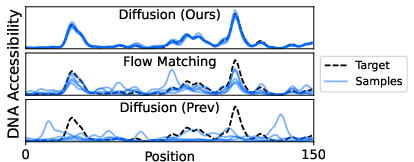
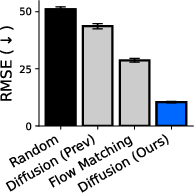
- Better DNA generation: With the stable simplicial approach, the model generates DNA sequences that match high-dimensional biological properties (like “accessibility profiles”) much more accurately than previous diffusion models. Their ELBO (a training score) improves dramatically, and conditional samples better match targets.
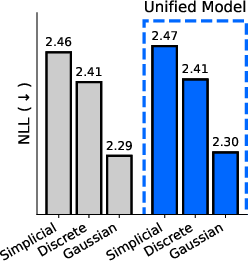
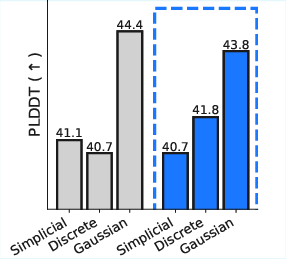
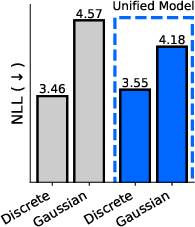
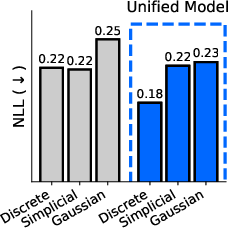
- One model for all domains: They introduce the “sufficient statistic parameterization” (SSP). It boils down to feeding the model a standard summary of the noisy evidence at each position that works the same way for all three diffusion types. As a result, they train a single network that can switch between discrete, Gaussian, and simplicial diffusion at test time without retraining. In experiments on proteins and language, this unified model matches or competes with separate specialist models trained individually.
What could this change?
- Easier model choice: You don’t need to pick one diffusion type before training. A single, unified model can handle multiple domains and tasks, widening the toolbox for real-world applications.
- Fair comparisons: Likelihoods across different diffusion families can be compared on equal footing. This helps researchers decide which approach is best for a given dataset or goal.
- More stable algorithms: Simplicial diffusion becomes fast and reliable, opening doors to better biological sequence design and other tasks that need probability distributions over symbols.
- Better transfer and tuning: The SSP idea can extend beyond just three diffusion types. It could let you train across different hyperparameters, switch schedules, or even adapt to new modalities—without starting from scratch.
In short, the paper builds a solid bridge between three popular diffusion methods, fixes a major stability issue, and shows you can train one versatile model that works across domains. This makes diffusion modeling simpler to use, more comparable, and more powerful for science and engineering.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
The following points identify what is missing, uncertain, or left unexplored in the paper, framed to enable concrete follow-up work:
- Formal unification beyond parent-independent mutation
- Extend the Wright–Fisher (WF) unification to parent-dependent mutation, selection (non-neutral WF), recombination, and variable population size, and derive the corresponding forward process and ELBO.
- Clarify whether the Jenkins exact-simulation and the proposed ELBO generalize to these richer genetic models.
- Conditions and edge cases for the Gaussian limit
- Specify the full regularity conditions on the rate matrix (e.g., non-diagonalizable generators, defective Jordan forms, degenerate or multiple slow eigenvalues, complex eigenpairs) under which the projection onto the first eigenspace and the Gaussian limit are valid.
- Analyze cases with multiple equally slow eigenmodes (eigenvalue multiplicity) and the impact on the embedding rank and downstream training.
- Independence across sequence positions
- The theory and training assume independent forward processes per position; develop a unified forward process and ELBO that explicitly model inter-position dependencies (e.g., motifs, structural couplings in proteins, syntax in language) rather than relying solely on the neural decoder to capture them.
- Scaling simplicial diffusion to large alphabets
- Provide algorithms and complexity analysis to scale WF simplicial diffusion (exact sampling and score computation) to very large vocabularies (e.g., language with ), including memory and time considerations and stability guarantees.
- Low-t approximations and stability guarantees
- The stabilization relies on heuristic thresholds (e.g., ) and CLT-based approximations; derive error bounds, adaptive switching strategies, and training-time diagnostics that guarantee numerical stability without degrading gradients.
- Quantify how the low- approximations affect the variance and bias of the ELBO estimator during training.
- Hollow parameterization: scope and side effects
- Establish the precise conditions under which the hollow parameterization yields formally comparable ELBOs across modalities; delineate cases where comparability fails.
- Study its effect on sample diversity, calibration, and bias–variance trade-offs, and whether analogous constructions exist for continuous domains beyond discrete tokens.
- Hyperparameter learning and design
- Move from hand-designed (or embeddings) to data-driven estimation: propose procedures to learn (including its eigenstructure) jointly with the model or via meta-learning, and quantify how errors in propagate to the unified embeddings and ELBO.
- Provide practical guidelines to design to target a desired embedding dimension and structure, with interpretability checks beyond qualitative plots.
- ELBO for simplicial diffusion: tightness and variance
- Prove tightness properties (or bounds) of the derived ELBO for WF simplicial diffusion versus the heuristic score-matching objective, and analyze estimator variance under realistic training regimes.
- Investigate alternative metrics (beyond the Fisher geometry diag) and their impact on optimization and sample quality.
- Unified guidance and inference procedures
- Systematically develop and benchmark classifier guidance, conditional sampling, and other diffusion-time inference algorithms across all three domains within the unified framework (including under SSP), with ablation on guidance strength and stability.
- SSP generalization and transfer
- Provide theoretical guarantees (or counterexamples) for SSP’s ability to generalize to unseen modalities or hyperparameter settings, and characterize preconditions (e.g., accurate forward marginals ) needed for transfer.
- Investigate catastrophic forgetting and curriculum strategies when alternating modality-specific ELBOs during training; quantify trade-offs vs single-modality specialization.
- Empirical breadth and metrics
- Expand empirical evaluation beyond the current datasets: test on large-scale images, diverse protein families, multiple language corpora, and additional biological assays.
- Standardize reporting (ELBO vs NLL, sign conventions) and provide cross-modal calibration procedures to ensure meaningful likelihood comparisons.
- Flow matching, masking, reflected diffusion, and indel processes
- Formalize the connections (proofs, objective mappings, and likelihood comparability) to flow matching, masking diffusion, reflected diffusion, and insertion–deletion processes; provide unified sampling and training recipes.
- Boundary behavior and degeneracies on the simplex
- Analyze and mitigate numerical pathologies near the simplex boundaries (e.g., coordinates near 0 or 1, degenerate ), including regularization strategies and their theoretical implications.
- Computational trade-offs and parallelization
- Quantify speedups vs SDE simulation for WF exact sampling, provide parallel implementations (GPU/TPU), and characterize memory and throughput when scaling sequence length and batch size.
- Practical design space between modalities
- The theory suggests a continuum of models between discrete, Gaussian, and simplicial diffusion; define and implement intermediate forward processes and assess whether they offer superior trade-offs for specific tasks.
- Biological validation of generated sequences
- Move beyond model-based surrogates (e.g., accessibility predictors, pLDDT) to experimental validation of DNA/protein designs; assess functional viability, off-target effects, and robustness.
- Robustness to schedule choices
- Investigate sensitivity to the time-dilation function across modalities, propose adaptive or learned schedules, and examine their effect on ELBO comparability and sample quality.
- Guidance on choosing modality at inference
- Provide decision procedures (based on task characteristics and resource constraints) for selecting the most appropriate domain (discrete, Gaussian, simplicial) when using a unified model at test time.
- Reproducibility and ablation transparency
- Supply detailed ablations on each stabilization component (exact sampling vs SDE, low- approximation, metric choice, hollow parameterization, SSP), with open benchmarks to facilitate replication and extension.
Practical Applications
Immediate Applications
The paper’s unification of discrete, Gaussian, and simplicial diffusion via the Wright–Fisher (WF) framework, plus its stability and training innovations, enable several deployable use cases and workflows:
- Healthcare/Biotech — conditional DNA design and optimization
- Use cases:
- Design regulatory DNA (promoters/enhancers) conditioned on target accessibility profiles for cell-type specific expression.
- Optimize CRISPR guide or donor sequences for predicted on-target efficacy while minimizing off-target profiles.
- Rapid “property-guided” sequence generation for synthetic biology (e.g., TF binding, methylation profiles).
- Tools/workflows:
- Stable Wright–Fisher simplicial diffusion with exact marginal sampling (Jenkins algorithm) and low‑t stabilization for fast, numerically robust training and inference.
- Classifier-guided sampling (e.g., accessibility predictors) plugged into diffusion workflows.
- Likelihood-based model selection using comparable ELBOs across modalities (enabled by the hollow parameterization).
- Assumptions/dependencies:
- Availability and quality of sequence–property predictors (e.g., accessibility, binding, expression).
- Parent-independent mutation setting (π, ψ) for WF-based simplex diffusion; independence across positions during forward corruption.
- Hollow parameterization is used for loss comparability; codebase and data access.
- Protein engineering — unified training and generative design
- Use cases:
- Generate foldable protein sequences conditioned on structural plausibility (e.g., pLDDT or design constraints).
- Use one model to switch modalities (discrete, Gaussian, simplicial) for downstream sampling strategies without retraining.
- Tools/workflows:
- Sufficient Statistic Parameterization (SSP) to train a single network that generalizes across diffusion domains; alternating ELBO training across modalities.
- Embedding sanity-check via mapping from mutation matrix L to the dominant eigenspace (visual inspection of amino acid clusters).
- Assumptions/dependencies:
- Access to structure predictors (AlphaFold-like), curated protein datasets.
- Dominant eigenspace captures key sequence variation; target distribution p(x0) fixed during SSP training.
- Software/ML — model benchmarking, selection, and deployment
- Use cases:
- Compare and select diffusion models on discrete data using formally comparable likelihoods (ELBOs), removing ambiguity in evaluation.
- Reduce operational complexity by “train once, deploy on many” with SSP; swap diffusion domain at inference based on task needs.
- Improve stability and speed of simplex diffusion by replacing SDE simulation with exact marginal sampling (Jenkins), plus low‑t fixes.
- Tools/workflows:
- “Model chooser” pipeline: compute comparable ELBOs across discrete/continuous/simplex variants; pick best domain for a task.
- “Low‑t stabilizer” patch for simplicial diffusion (central limit approximation for A(ψ, τ)).
- Architecture-agnostic hollow predictor: qθ(x0d | xt, t) ∝ p(xtd | x0d, t) qθ(x0d | xt−d, t) to remove singularities and improve early-stage training.
- Assumptions/dependencies:
- Consistency in p(x0) across training when applying SSP; schedule conditioning handled via sufficient statistics φ.
- Data quality and adequate compute for multi-domain training.
- Language technology — improved generative modeling and controllability
- Use cases:
- Train a single diffusion-based LLM that can run in discrete or Gaussian embedding space (e.g., for different sampling/conditioning strategies).
- Better perplexity and sample quality without fragile schedule choices (SSP makes models more time-invariant).
- Tools/workflows:
- SSP-based unified training for text; hollow parameterization to stabilize early denoising steps.
- Embedding design guided by L→embedding mapping to make token similarity structure explicit.
- Assumptions/dependencies:
- Large vocabulary size limits current simplex scaling; discrete/Gaussian are immediately applicable.
- Policy and risk governance — responsible synthetic sequence generation
- Use cases:
- Institutional review pipelines that integrate stronger generative capability from stable simplex diffusion for DNA, with pre-deployment risk assessments.
- Auditable comparisons across models using ELBOs to enforce consistent benchmarking in regulated settings.
- Tools/workflows:
- Governance checklists aligned to unified diffusion tooling (e.g., gated release, controlled property predictors, misuse monitoring).
- Assumptions/dependencies:
- Organizational biosecurity policies; access controls and model cards documenting capabilities and limitations.
- Daily life/consumer apps — higher-quality text generation
- Use cases:
- Integrate unified diffusion LLMs into content creation apps for improved fluency and controllability (e.g., stylized generation via domain switching).
- Tools/workflows:
- SSP-based training to reduce sensitivity to schedules; deployment that toggles discrete vs Gaussian diffusion based on latency/quality trade-offs.
- Assumptions/dependencies:
- Adequate inference infrastructure; safeguards for content quality and safety.
Long-Term Applications
The framework opens pathways that need scaling, additional research, or productization before broad deployment:
- Cross-domain foundation models for discrete sequences (biology, language, other categorical streams)
- Vision:
- Train SSP-based universal sequence generators that transfer across domains and tasks (e.g., DNA→protein→text) without retraining.
- Potential products:
- “Diffusion-on-demand” SDK that selects modality and hyperparameters at inference time; plug-in property predictors.
- Assumptions/dependencies:
- Robust domain adaptation; shared or compatible p(x0); strong property predictors across sectors.
- Hyperparameter-on-the-fly tuning and schedule invariance
- Vision:
- Use SSP to train once and then vary time schedules, mutation matrices L, or embeddings at inference without retraining.
- Potential products:
- Auto-hyperparameter planner that maps desired inductive biases (via L or embedding space) into model behavior for specific tasks.
- Assumptions/dependencies:
- Validity of sufficient-statistic reduction across new hyperparameter regimes; careful calibration of φ mappings.
- Scalable simplicial diffusion for large vocabularies (e.g., full-scale language)
- Vision:
- Extend WF-based simplex methods (exact marginals, stabilized low‑t loss) to B ≈ 104–105 tokens with efficient algorithms.
- Potential products:
- Simplex-native LLMs supporting closed-form likelihoods and guidance; better controllability over categorical mixtures.
- Assumptions/dependencies:
- Algorithmic innovations for high-dimensional Dirichlet and series approximations; memory/runtime optimization.
- Unified diffusion–flow matching frameworks
- Vision:
- Bridge closed-form ELBO and guidance from diffusion with stability and scalability from flow matching on the simplex.
- Potential products:
- Hybrid training stacks that preserve likelihood computation and sampling guidance while retaining the speed and robustness of flows.
- Assumptions/dependencies:
- New theory to reconcile objectives and sampling; practical gradient estimators without prohibitive cost.
- Intermediate models between discrete, Gaussian, and simplex
- Vision:
- Exploit WF parameters (population size ζ, reproduction rate, mutation structure) to craft task-specific intermediate diffusions.
- Potential products:
- Domain-tuned generators (e.g., partial reproduction for semi-categorical streams in healthcare telemetry, IoT event sequences).
- Assumptions/dependencies:
- Careful analysis of eigenspace behavior; validation on domain data.
- Robotics and controls — generative action sequence planning
- Vision:
- Use unified diffusion for categorical action sequences with property-guided objectives (e.g., safety or energy efficiency).
- Potential products:
- Planner modules that switch modalities for exploration vs exploitation; likelihood-based evaluation across planners.
- Assumptions/dependencies:
- Accurate property predictors in robotics; task-specific constraints; safe deployment.
- Energy and finance — event-stream simulation and stress testing
- Vision:
- Model categorical event sequences (grid events, order book states) using stable simplex diffusion with direct likelihoods.
- Potential products:
- Scenario generators for risk analysis that can be guided by constraints (e.g., rare-event targeting) using classifier guidance.
- Assumptions/dependencies:
- Domain data quality; calibrated classifiers; regulatory alignment.
- Policy — standardized evaluation and governance for generative biology
- Vision:
- Use ELBO comparability and unified tooling to define benchmarks, release criteria, and safe deployment practices for synthetic sequence models.
- Potential products:
- Auditing frameworks and dashboards that visualize embedding structures (from L), capability reports, and misuse controls.
- Assumptions/dependencies:
- Multistakeholder consensus; integration with existing biosafety standards.
Each application’s feasibility depends on key assumptions highlighted in the paper: the hollow parameterization for loss comparability; SSP requiring a fixed target distribution p(x0); independence across sequence positions during forward corruption; valid parent-independent mutation settings for WF simplex diffusion; availability of accurate property predictors; and sufficient compute/data to realize multi-domain training.
Glossary
- BLOSUM: A family of amino acid substitution matrices used in bioinformatics to quantify similarity or substitution probabilities between residues. "for the BLOSUM stochastic processes for amino acids"
- Brownian motion: A continuous-time Gaussian process modeling random motion; used as the forward noise process in Gaussian diffusion. "the forward process is Brownian motion on embeddings"
- Central limit approximation: An approximation that replaces a distribution with a normal distribution in certain regimes, improving numerical stability at small times. "with a central limit approximation for "
- Central limit theorem: A theorem stating that averages of independent random variables converge to a normal distribution under broad conditions. "because of the central limit theorem"
- Classifier guidance: A sampling technique that steers diffusion models using gradients from an auxiliary classifier to achieve desired properties. "such as classifier guidance."
- Cox–Ingersoll–Ross process: A stochastic process from mathematical finance with nonnegative state space; used here as a building block for simplex diffusion. "the ``Cox-Ingersoll-Ross process''"
- Dirichlet distribution: A probability distribution over the simplex, often used as a prior for categorical probabilities. "Sample "
- ELBO (Evidence Lower Bound): A variational objective that lower-bounds the log-likelihood, optimized to train diffusion models. "optimize a negative ELBO"
- Eigenspace (left eigenspace): The subspace spanned by eigenvectors associated with a specific eigenvalue of a matrix, taken with respect to left multiplication. "projection onto the left eigenspace"
- Eigenvalues: Scalars associated with a linear transformation that scale eigenvectors; here, they govern decay rates of modes in the rate matrix. "the eigenvalues of "
- Flow matching: A generative modeling framework that learns vector fields (flows) to map simple distributions to data distributions, here applied on the simplex. "build flow-matching models on the simplex"
- Gaussian diffusion: A diffusion modeling approach that adds Gaussian noise in a continuous embedding space. "Gaussian diffusion has more mature sampling and training procedures"
- Hollow parameterization: A parameterization that weights model predictions by the likelihood of observations given hypotheses, improving stability and comparability of likelihoods. "which we call the hollow parameterization."
- Infinitesimal flow: The instantaneous transition dynamics of a process; in diffusion training, it refers to the local forward and backward probability flows. "the ``infinitesimal flow'' forward and backward "
- Jacobi process: A diffusion process on the simplex obtained by normalizing certain positive processes, used for simplicial diffusion. "the ``Jacobi process''"
- Kullback–Leibler (KL) divergence: A measure of how one probability distribution diverges from another. "is the KL divergence between two Poisson distributions"
- Law of large numbers: A theorem stating that averages of independent random variables converge to their expected value. "by the law of large numbers,"
- Masking diffusion: A discrete diffusion variant where tokens are progressively masked; known for time-invariance properties. "time-invariance of masking diffusion"
- Parent-independent mutation rate matrix: A mutation generator where mutation rates depend only on the target state distribution and not on the current parent state. "a parent-independent mutation rate matrix"
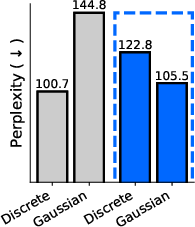
- Perplexity: An evaluation metric in language modeling that measures how well a probability model predicts a sample. "measure sample quality using the perplexity of a much larger LLM."
- pLDDT: A metric from protein structure prediction assessing per-residue confidence, used to evaluate generated protein quality. "measure sample quality by predicted protein fold-ability (pLDDT)."
- Rate matrix (mutation rate matrix): The infinitesimal generator of a continuous-time Markov process describing mutation rates between tokens. "the forward process is mutation defined with a rate matrix "
- Score function: The gradient of the log-density with respect to inputs; matching score functions is used as a loss for diffusion on the simplex. "matches ``score functions'' "
- SDE (stochastic differential equation): A differential equation driven by stochastic processes (e.g., Brownian motion), used to simulate diffusion dynamics. "a stochastic differential equation (SDE)"
- Simplex: The space of nonnegative vectors that sum to one; the natural domain for distributions over categorical probabilities. "diffusion on a simplex"
- Simplicial diffusion: Diffusion processes defined on the probability simplex, intended to blend discrete naturalness with continuous algorithms. "simplicial diffusion models"
- Stationary distribution: A distribution that remains invariant under the evolution of a Markov process. "a stationary distribution "
- Stationary mutation distribution: The stationary distribution induced by the mutation rate matrix, around which Dirichlet samples are centered. "centred at the stationary mutation distribution "
- Stirling's approximation: An approximation for factorials (or gamma functions) used to simplify expressions in asymptotic analysis. "Stirling's approximation"
- Sufficient statistic parameterization (SSP): A parameterization where inputs are reduced to sufficient statistics, enabling a single model to operate across multiple diffusion modalities. "The sufficient statistic parameterization (SSP)"
- Time dilation function: A monotone mapping that re-parameterizes diffusion time to align training and stationary distributions. "The time dilation function"
- Time-invariance: A property where predictions do not depend on the specific diffusion time when expressed in sufficient statistics. "the noted ``time-invariance'' of masking diffusion"
- Wright–Fisher diffusion: The diffusion limit on the simplex arising from the Wright–Fisher model as population size tends to infinity. "Wright-Fisher diffusion with population size "
- Wright–Fisher model: A classical population genetics model describing mutation and reproduction in finite populations, unifying discrete, Gaussian, and simplicial diffusion. "the Wright-Fisher (WF) model."
Collections
Sign up for free to add this paper to one or more collections.