- The paper shows that activity prediction can be confounded by lab-specific stylistic signals rather than true chemical causality.
- It employs an author–molecule framework using ECFP descriptors and gradient boosting, achieving a notable top-5 accuracy of 60% on a scaffold-disjoint split.
- The findings advocate for source-aware benchmarking with lab-disjoint splits and metadata retention to mitigate shortcut learning in SAR models.
Chemist Style as a Confounder in Medicinal Chemistry Benchmarks
Introduction
The paper "Clever Hans in Chemistry: Chemist Style Signals Confound Activity Prediction on Public Benchmarks" (2512.20924) critically examines the influence of chemist-specific stylistic signals on the predictive performance of ML models in structure-activity relationship (SAR) tasks leveraging public datasets such as ChEMBL. By establishing a robust author–molecule framework, the authors demonstrate that bioactivity prediction can largely exploit regularities rooted in laboratory practices, effectively confounding the attribution of predictive power to true chemical causality.
Shortcut Learning and the Clever Hans Phenomenon
Shortcut learning in computer vision has been extensively documented, where spurious dataset-specific signals—distinct from causal mechanisms—drive model decisions and undermine out-of-distribution generalization. The "Clever Hans" metaphor typifies this phenomenon, where an ML model exploits a superficial cue (e.g., a photographer's watermark in horse images) rather than the intended semantic content. In the context of ChEMBL assays, analogous shortcuts emerge: models trained on literature-sourced compound collections capture laboratory-specific design motifs, reaction schemes, and target portfolios, instead of molecular determinants of bioactivity.

Figure 1: Classic example of a Clever Hans predictor—relevance heatmap focus on a watermark rather than on the horse, illustrating a non-causal shortcut.
Author Identification: Quantifying Chemist Style
The authors construct a detailed mapping of molecules to publication authors in ChEMBL, yielding 1,815 prolific author classes. Using ECFP-based molecular fingerprints and a gradient boosting classifier (LightGBM), they assess the learnability and distinctiveness of "chemist style" by predicting the originating author from structure alone. The model attains a top-5 accuracy of 60% in a scaffold-disjoint split, an order of magnitude above chance, substantiating the existence of highly separable stylistic domains within medicinal chemistry.
Analysis of one-vs-rest ROC-AUC distributions reveals pronounced heterogeneity across authors. Some chemists yield highly distinctive structural signatures traceable to unique scaffolds and synthetic strategies, while others present less discriminable patterns.
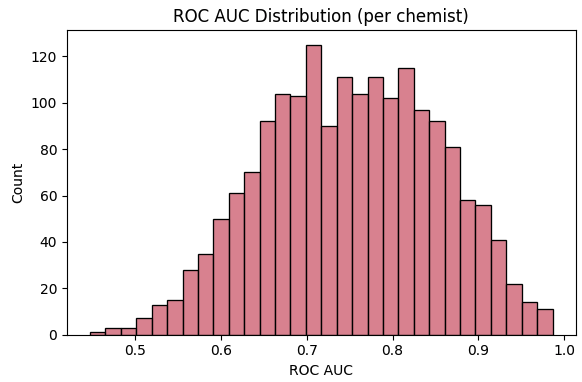
Figure 2: Histogram of author-specific ROC-AUC scores in binary author classifiers—displaying the wide variation in stylistic identifiability.
Case studies of top-performing author predictions show that certain chemists, such as Carrie Haskell-Luevano, T. Harrison, and Brian S. J. Blagg, are unambiguously identifiable by stylized molecular features. Their confusion matrices exhibit sharp diagonal dominance, with off-diagonal errors primarily to chemically similar labs.
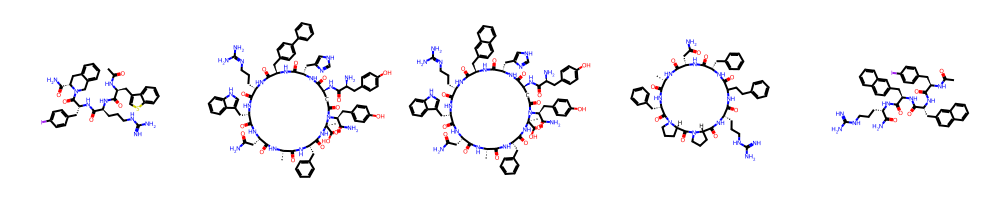
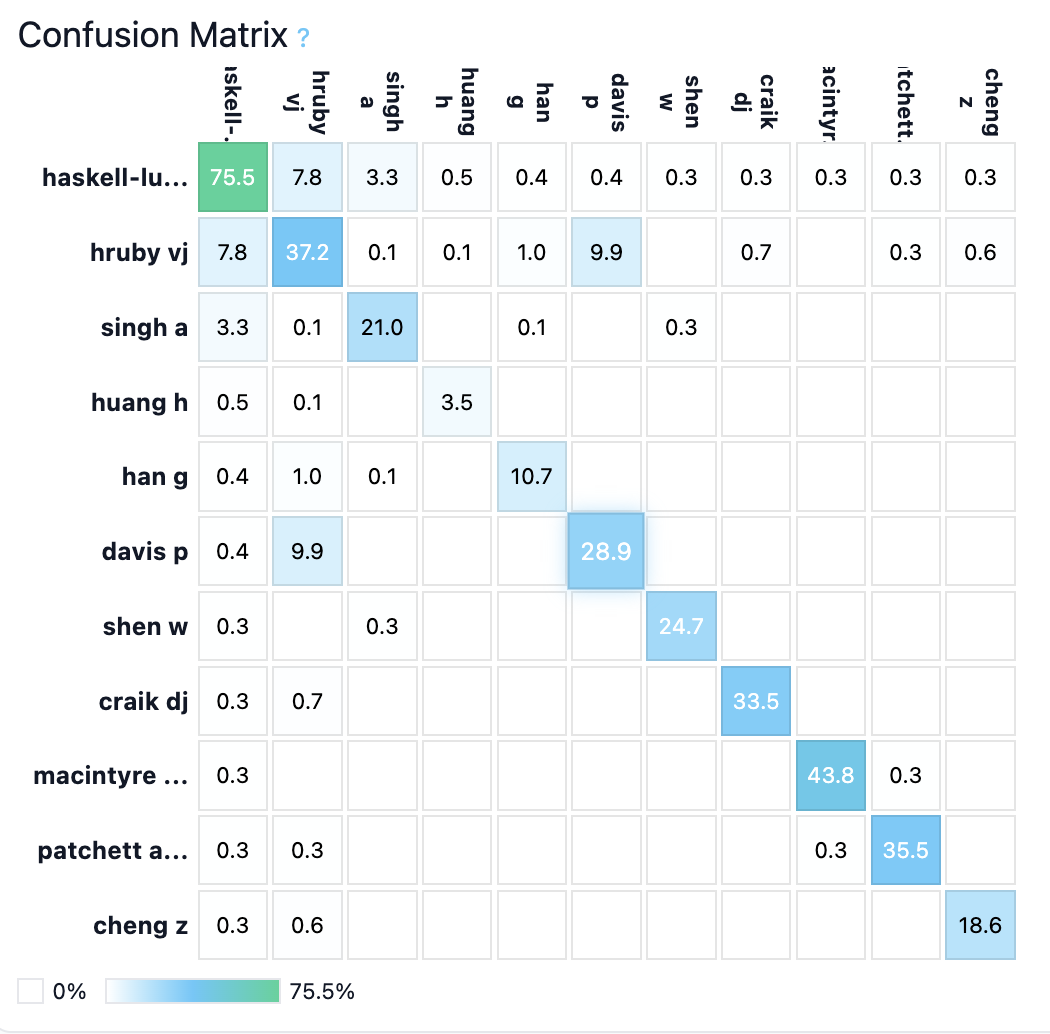
Figure 3: Haskell-Luevano exemplars and confusion profile—clearly demarcated chemist style in macrocyclic ligand series.
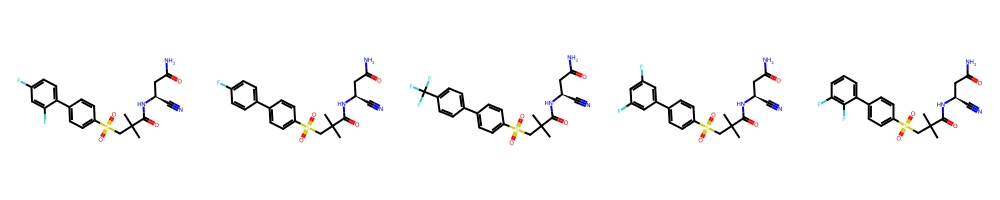
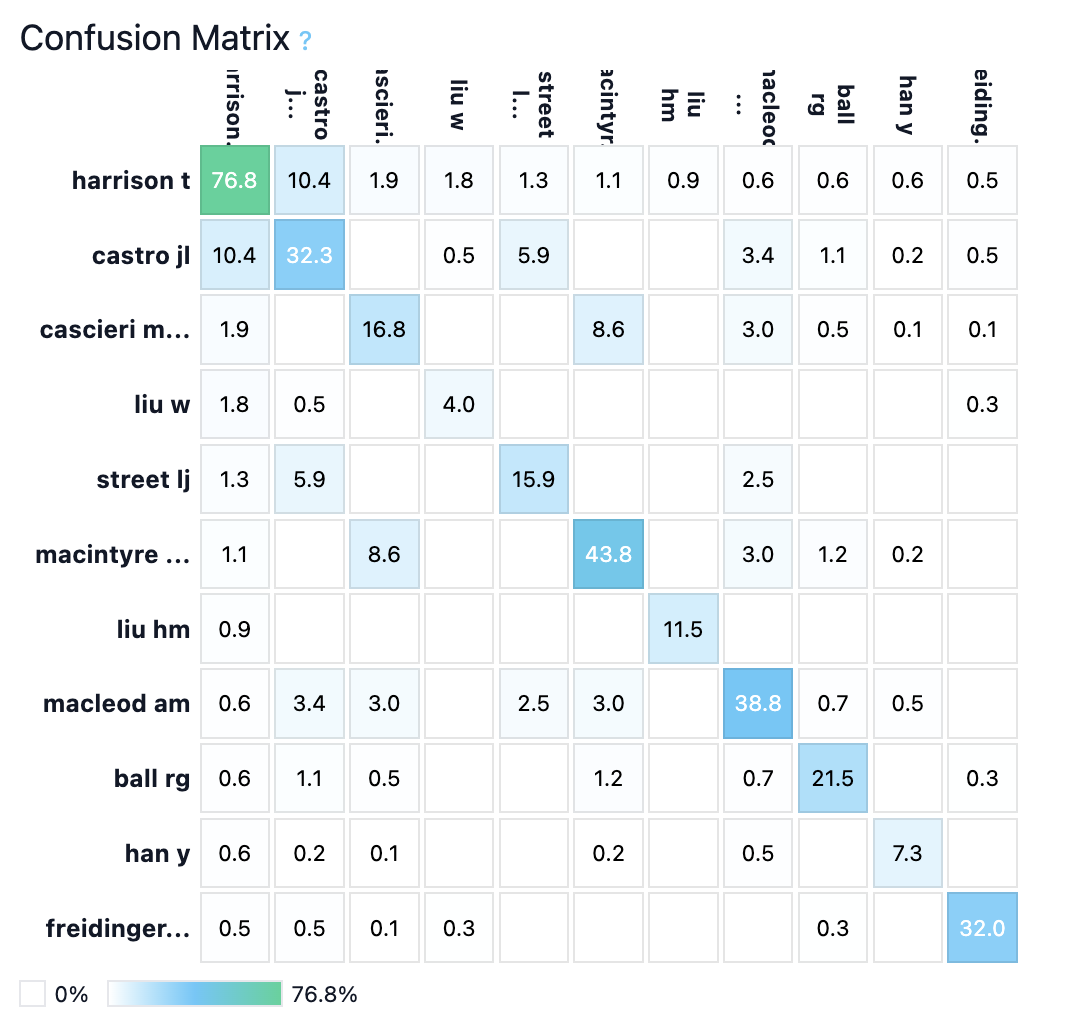
Figure 4: Harrison exemplars and confusion matrix—SAR series with distinctive heterocycles yield highly separable style.
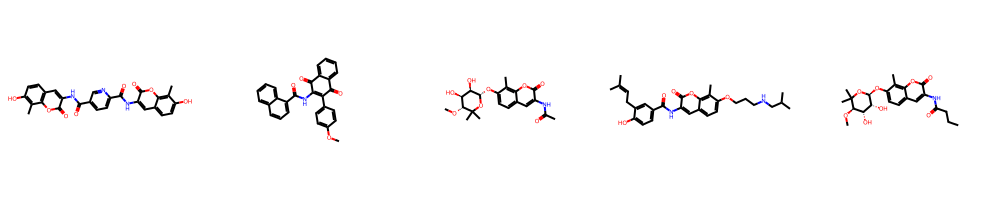
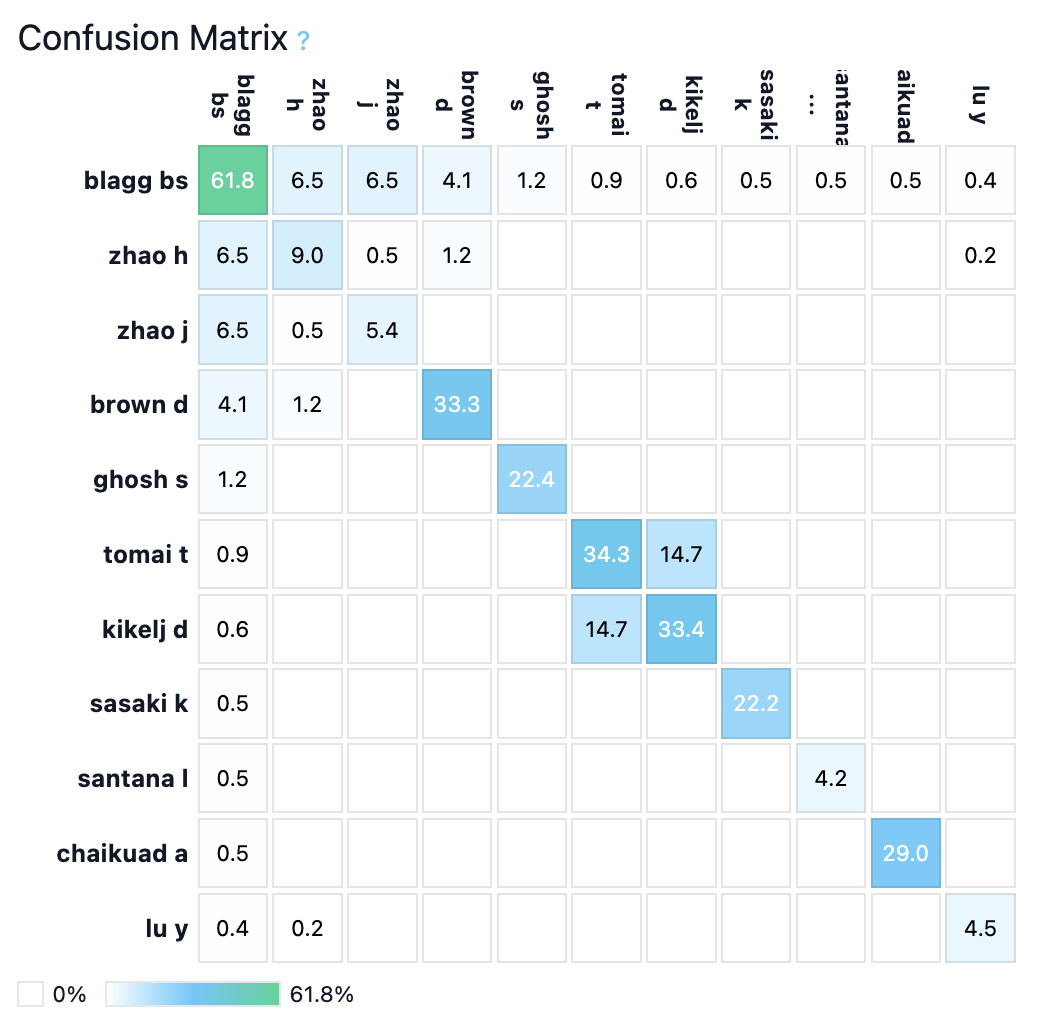
Figure 5: Blagg exemplars and confusion matrix—natural product–derived analogues with unique substitution patterns drive recognizability.
Activity Prediction: Intent Leakage as a Dominant Signal
The principal claim is demonstrated via a two-stage modeling framework: Stage 1, an author classifier generates a probability vector encapsulating chemist style for each molecule; Stage 2, an activity model receives this author-probability vector and protein ID only, devoid of molecular descriptors. The activity model thus operates solely on proxies for chemist intent and target identity.
Empirically, the author-probability + protein model achieves a mean validation AUROC of 0.652, closely matching a baseline ECFP + protein model (AUROC 0.656), with marginal gains when both representations are combined. The author-derived model approaches precision-recall performance adequate for practical use despite its structural agnosticism, contravening the notion that chemical structure per se underlies activity prediction on benchmark data.
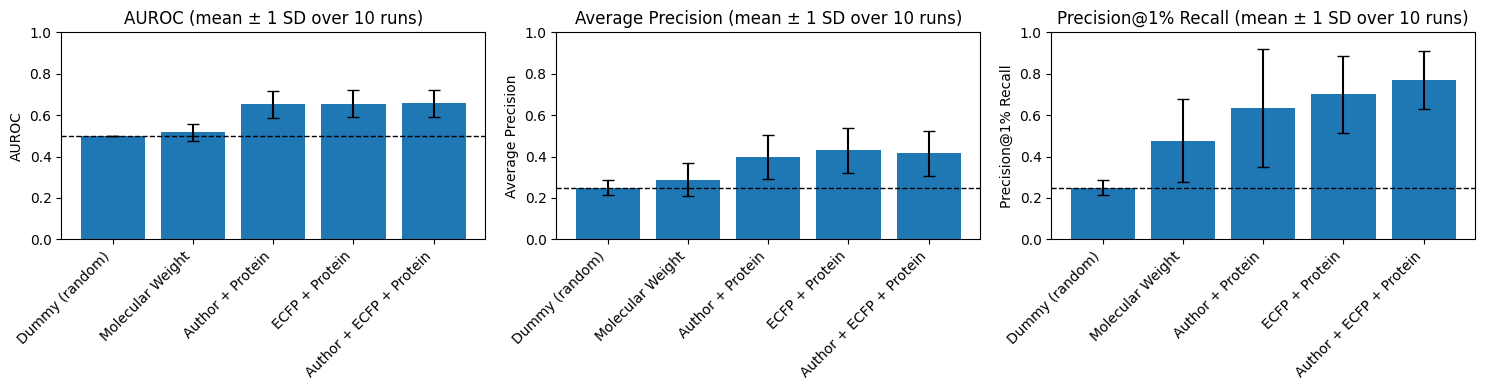
Figure 6: Validation performance comparison—author + protein and ECFP + protein models deliver commensurate AUROC and AP, revealing the predominance of chemist style and target identity.
Critically, the ability to decode molecular fingerprint bits from the author-probability vectors (median ROC-AUC ≈ 0.9) indicates that the representation not only captures intent proxies but also encodes substantial chemical information, albeit in a lab-centric embedding space.
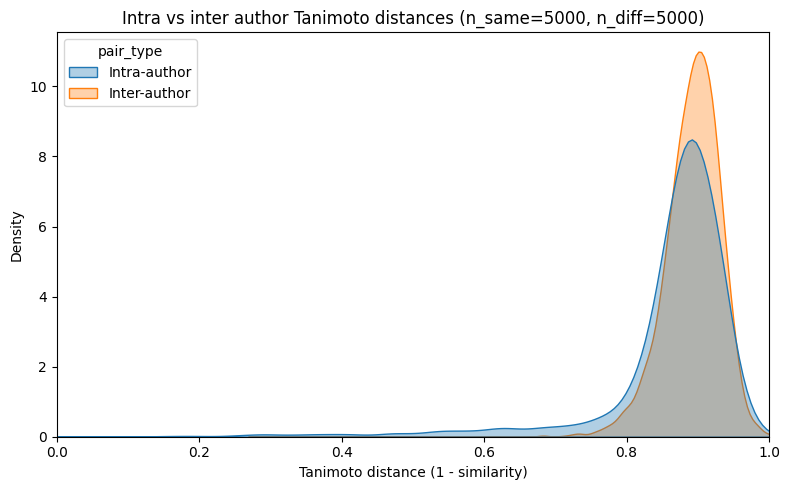
Figure 7: Tanimoto distance analysis—prediction ease does not arise from trivial inter/intra-author chemical similarity.
Implications and Recommendations
These findings underscore the necessity for source-aware benchmarking in cheminformatics. The entanglement of chemist style and data provenance with activity labels introduces a confound that artificially boosts model performance. Such shortcut exploitation raises concerns about generalizability and causal interpretability, with practical risks for prospective drug discovery—particularly for campaigns seeking scaffold/chemical novelty unconstrained by historical laboratory output.
To address these confounds, the paper advocates for:
- Retention and explicit reporting of source metadata (authors, labs, vendors) in datasets.
- Inclusion of source-only/source+target baselines to quantify non-causal predictive contributions.
- Adoption of author/lab/site-disjoint splits in addition to scaffold and temporal splits for robust evaluation.
These interventions are critical for delineating legitimate SAR learning versus dataset-specific intent leakage and for advancing trustworthy AI in medicinal chemistry.
Conclusion
The work rigorously demonstrates that author-linked signals pervade public medicinal chemistry datasets, enabling models to achieve strong activity prediction performance by inferring chemist intent and institutional regularities rather than learning transferable chemical biology mechanisms. This "Clever Hans" failure mode has substantial implications for model evaluation, deployment, and the design of future benchmarks. Theoretical progress will require systematic mitigation of provenance-based confounds, while practical applications must consider the limitations of shortcut-driven generalization when extrapolating beyond well-trodden chemical space.