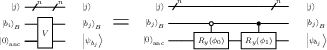Quantum Elastic Network Models and their Application to Graphene
Published 8 Jan 2026 in quant-ph, cond-mat.mtrl-sci, and physics.comp-ph | (2601.05161v1)
Abstract: Molecular dynamics simulations are a central computational methodology in materials design for relating atomic composition to mechanical properties. However, simulating materials with atomic-level resolution on a macroscopic scale is infeasible on current classical hardware, even when using the simplest elastic network models (ENMs) that represent molecular vibrations as a network of coupled oscillators. To address this issue, we introduce Quantum Elastic Network Models (QENMs) and utilize the quantum algorithm of Babbush et al. (PRX, 2023), which offers an exponential advantage when simulating systems of coupled oscillators under some specific conditions and assumptions. Here, we demonstrate how our method enables the efficient simulation of planar materials. As an example, we apply our algorithm to the task of simulating a 2D graphene sheet. We analyze the exact complexity for initial-state preparation, Hamiltonian simulation, and measurement of this material, and provide two real-world applications: heat transfer and the out-of-plane rippling effect. We estimate that an atomistic simulation of a graphene sheet on the centimeter scale, classically requiring hundreds of petabytes of memory and prohibitive runtimes, could be encoded and simulated with as few as $\sim 160$ logical qubits.
The paper introduces QENMs that efficiently map coupled oscillators into quantum states, enabling simulation of macroscopic graphene with only ~160 logical qubits.
It develops state preparation methods using discretized Maxwell-Boltzmann distributions and optimized oracles that leverage graphene’s lattice symmetry.
Resource analysis demonstrates exponential memory savings and practical polynomial time complexity for simulating a square centimeter of graphene.
Quantum Elastic Network Models and Their Application to Graphene
Overview of Quantum Elastic Network Models (QENMs)
This work introduces Quantum Elastic Network Models (QENMs) as a quantum computational extension of classical Elastic Network Models (ENMs), targeting efficient simulation of vibrational dynamics in large-scale molecular systems. Utilizing the quantum algorithmic framework of Babbush et al. (2023), QENMs encode networks of coupled harmonic oscillators—commonly used to simulate biomolecules and materials—directly onto quantum states. Of particular emphasis is the demonstration that planar materials such as graphene, whose atomistic representation at macroscopic scales is intractable for current classical hardware, can be efficiently handled with quantum resources scaling logarithmically in system size under favorable conditions.
The approach starts from the ENM formalism, where nodes (atoms or coarse-grained units) are connected by harmonic springs if within a spatial cutoff, resulting in sparse connectivity matrices. Through mapping the Newtonian dynamics of these oscillators to the Schrödinger equation, QENMs leverage block-encoded Hamiltonian simulation, space-efficient amplitude encoding, and structured quantum oracles to simulate system evolution and extract physical observables. The algorithmic complexity, resource estimates, and quantum state preparation procedures are investigated in detail. Significant focus is given to the construction of efficient oracles for molecules with high spatial structure, such as crystalline graphene, allowing the encoding of a square centimeter of graphene (3.8×10¹⁵ atoms) in approximately 160 logical qubits.
Classical ENMs and Mapping to Quantum Simulation
ENMs provide tractable mechanistic models for large biomolecular systems by substituting detailed force fields with harmonic interactions. Each oscillator represents an atom or group of atoms; connections are drawn for pairs within a cutoff radius, yielding a sparse network Laplacian with spring constants as weights. The quadratic potential enables efficient diagonalization but becomes infeasible for truly macroscopic systems due to exponential scaling in classical memory and runtime requirements.
The quantum mapping exploits a crucial observation: the equations of motion for a system of coupled oscillators can be encoded into quantum evolution under a corresponding Hermitian block-encoded Hamiltonian H. State preparation yields the initial positions and velocities, after which quantum simulation time-evolves the system, and observables (e.g., kinetic/potential energy, mean squared displacement) are estimated by projective measurements on evolved quantum states. The exponential space advantage arises because N=2n oscillators are encoded using only n qubits, with essential physical quantities extracted via amplitude estimation.
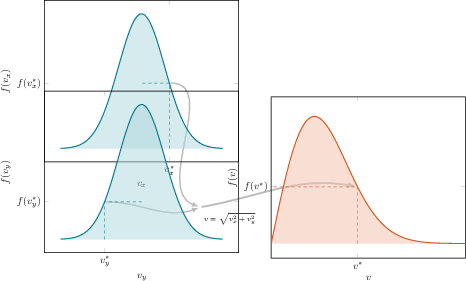
Figure 1: Sampling from the Boltzmann (Maxwell) velocity distribution via independent Gaussian sampling, foundational to initialization in realistic thermal simulations.
Efficient Quantum State Preparation
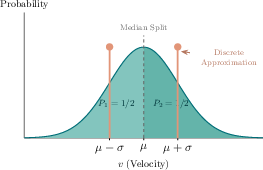
For faithful modeling of thermalized materials, initial atomic velocities must follow the Maxwell-Boltzmann distribution. Direct amplitude encoding of continuous distributions for large N is infeasible. This work proposes discretizing the Maxwell-Boltzmann distribution into a small number of equiprobable velocity buckets (e.g., k=2 for second-moment matching), enabling efficient, randomized assignment of bucketed velocities to atom indices.
Figure 2: Two-point approximation of the Maxwell-Boltzmann distribution: representative velocities and bucket probabilities strictly preserve mean and variance.
Figure 3: Quantum circuit for randomized bucket assignment; mapping node index j to a bucket bj via parity checks conditioned on a random string θ.
After bucket assignment, controlled rotations (using flag qubits) load the appropriate amplitude, corresponding to the selected velocity, into the quantum register:
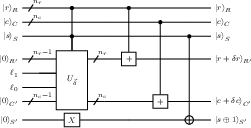
Figure 4: Velocity amplitude encoding circuit, where bucket flag qubits select the rotation angles mapping velocity amplitudes into ancilla states.
This preparation method remains efficient even when loading exponentially large numbers of velocities, due to leveraging pseudorandom functions and in-place logic rather than explicit memory-intensive storage or lookup.
Efficient Oracle Construction Leveraging Molecular Structure
A universal quantum advantage is conditional on the ability to construct all algorithmic oracles efficiently. For systems such as graphene, which feature translational and topological symmetry, the mass and connectivity oracles can be succinctly implemented using modular arithmetic and lattice coordinate decoding.
The connectivity oracle for graphene is carefully engineered by decomposing the lattice into a grid of unit cells, each with sublattice identifiers, and utilizing relative neighbor shifts plus boundary padding to distinguish physical from dummy (ghost) bonds. Quantum arithmetic circuits generate neighbor coordinates, and bond validity is enforced via logic circuits detecting out-of-bounds addresses.
Figure 5: Quantum circuit for neighbor computation via modular addition of relative shift vectors and sublattice toggling, central to efficient lattice traversal.
This eliminates costly QRAM lookups, ensuring resource demands scale logarithmically with system size.
Hamiltonian Simulation and Resource Analysis
Hamiltonian simulation utilizes a block-encoding of the system Laplacian. The sparsity d of graphene (each atom having three neighbors in the bulk) allows for optimized oracle calls and controls the scaling of the simulation subroutines. For homogeneous spring constants and masses, the construction of the Hamiltonian and ancillary matrices is simplified, and the block-encoded operator acts efficiently on an extended quantum space.
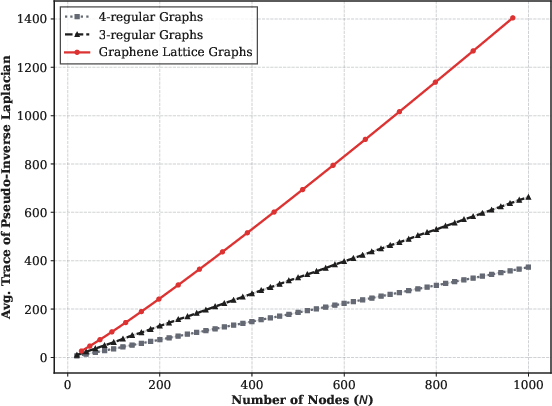
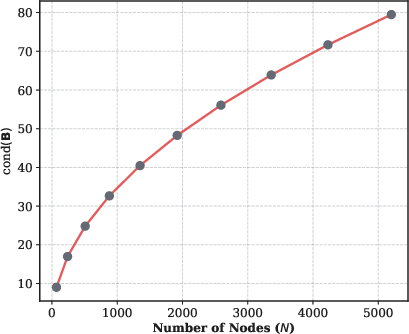
The alternative initial state encoding, leveraging the Moore-Penrose pseudo-inverse and projector onto the orthogonal null space of the Laplacian, enables direct extraction of node displacements, crucial for measuring physical observables (e.g., mean squared displacement, B-factors). The complexity of state preparation in this encoding is governed by the condition number of the sparse incidence matrix B, which, for regular graphs such as graphene, scales as O(N1/2), yielding O~(NlogN) overall cost.
Figure 6: Scaling of the trace of the pseudo-inverse Laplacian, showing near-linear behavior in system size for regular and graphene-like lattices.
Figure 7: The condition number of the incidence matrix B grows as O(N), reflecting polynomial overhead for displacement-accessible encoding in large systems.
Quantum signal processing and related Hamiltonian simulation primitives ensure that time evolution of the block-encoded system can be approximated to error ϵ with computational resources scaling favorably in both t and n=logN.
Measurement Protocols and Physical Applications
Limitations in projective quantum measurement mean that only observables with substantial probability amplitude—such as global energies or sufficiently large clusters—can be efficiently extracted; the effect of exponential suppression in local observables must be managed by careful experimental design. The methodology supports global kinetic/potential energy measurement (for heat flow and thermalization studies) as well as displacement-based quantities accessible via tailored encodings.
Key physical applications discussed include:
In-plane heat transfer: Preparation of localized thermal gradients followed by runtime tracking of kinetic energy distribution, exploiting binary partitioning strategies for spatial resolution. For truly macroscopic sheets (e.g., 1 cm²), exponential advantage emerges in space but time complexity becomes polynomial for long-time propagation.
Out-of-plane rippling (MSD dynamics): Measurement of time-averaged mean squared displacement along the z-direction, directly relating to experimentally observed rippling phenomena and macroscopic elastic properties. Though the quantum speedup is formally dequantized to a polynomial regime, high polynomial advantage persists for large enough system sizes.
Theoretical and Practical Implications
Theoretical Impact: This work clarifies the regimes in which QENMs (and more generally, quantum algorithms for oscillator networks) provide super-polynomial (or exponential) resource reduction over classical algorithms. Key to the quantum advantage is the presence of static structure, sparse connectivity, and efficient quantum encodings (both in amplitude and logical oracle circuits). The dequantization of short-time dynamics for locally connected networks, as proven in recent work, bounds expectations for absolute quantum advantage but does not invalidate the substantial reduction in resource scaling for medium- to long-time dynamics and exponentially large systems.
Practical Impact: The resource estimates show that quantum simulation of physically relevant macroscopic materials—previously requiring hundreds of petabytes—can become feasible with ∼160 logical qubits. This dramatically redefines the memory bottleneck in the simulation of atomistically detailed materials science problems. The present approach is well-suited for early fault-tolerant quantum hardware and can be readily adapted to other crystalline 2D materials given known symmetry and periodicity.
Extensions and Limitations: The formalism is presently restricted to harmonic (quadratic) potentials, which limits direct modeling of anharmonic effects, thermalization to experimental temperatures, and chemical reactions. Incorporating more realistic force fields (bonded/non-bonded, dihedral, and nonlinear interactions) remains a major challenge. Recent quantum algorithmic advances, such as nonlinear Schrödingerization techniques, suggest future generalizations are possible, potentially retaining quantum advantage for weakly nonlinear systems.
Conclusion
QENMs offer a robust and scalable framework for direct quantum simulation of large-scale elastic materials such as graphene, with clear exponential space savings and practical quantum resource requirements. While absolute quantum advantage is context- and observable-dependent, the presented methods enable substantial reductions in both space and time complexity compared to the best known classical alternatives under standard physical model assumptions. Future research will extend the methodology to support broader classes of interactions, incorporation of defects, non-periodic boundaries, and application to experimental conditions via thermal baths and open-system dynamics.