- The paper extends the CIPSI-driven CC(P;Q) approach to excited states, enabling near-CCSDT quality energetics via efficient selection of key triple excitations.
- The methodology partitions the excitation space into iterative (P) and non-iterative (Q) parts to drastically reduce computational cost while maintaining accuracy.
- Numerical benchmarks reveal that even minimal CIPSI-selected triple determinants can reduce errors to below 1.3 millihartree for multireference excited states, proving the method's robustness.
Extension of CIPSI-Driven CC(P;Q) Approach to Excited States
Introduction
The manuscript introduces a significant extension of the Coupled-Cluster Moment Expansion formalism, specifically the CIPSI-driven CC(P;Q) method, to the treatment of electronically excited states via the Equation-of-Motion Coupled Cluster (EOMCC) framework. The innovation leverages the Selected Configuration Interaction (CIPSI) method to identify the leading triply-excited determinants, enabling an efficient and accurate approach to approximate CCSDT/EOMCCSDT quality energetics with a substantially reduced computational footprint. This development addresses longstanding challenges in the accurate and efficient description of excited states, particularly those with substantial multireference diagnostic or strong double (and higher) excitation character that historically eluded lower-order EOMCCSD or perturbative triples corrections.
Methodology
The CC(P;Q) approach employs a partitioned working space: the P space (iterative zone) includes all singles and doubles plus a compact, CIPSI-selected subset of triple excitations, while the Q space comprises the remaining triples. In the ground and excited-state CC/EOMCC equations, the amplitudes corresponding to triple excitations are restricted to those appearing in P, resulting in substantial savings in both storage and computational cost. Non-iterative corrections—evaluated over the Q space using energy denominators of Epstein-Nesbet character—recover the missing triples contributions, heavily relying on the CC(P) reference.
For excited states, the formalism follows the EOMCC paradigm, where state-specific right and left eigenvectors are optimized in the truncated excitation manifold. The extension requires construction of P spaces from multiple relevant CIPSI runs targeting both ground and excited-state symmetries, ensuring that the triples manifold is tailored for each target state's character. This adaptive selection is essential for acceptable convergence rates toward parent EOMCCSDT results.
CIPSI-Driven Determinant Selection
CIPSI provides an efficient means of generating highly relevant subsets of determinants. The iterative expansion stops once a user-specified termination criterion—either threshold on second-order perturbative corrections or total determinant size—is met. This allows systematic investigation of convergence with respect to triples coverage, enabling robust assessment of the balance between efficiency and correlation recovery.
Numerical Results
Vertical and Adiabatic Excitations ($\ce{CH+}$, CH)
The method was validated on the $\ce{CH+}$ ion at equilibrium and stretched geometries, as well as the CH radical, using high-quality atomic basis sets and benchmarked against full EOMCCSDT values. The results demonstrate:
- EOMCC(P)/Ndet(in)=1 (standard EOMCCSD) yields large errors for multireference or high double-excitation character states, e.g., up to 144 millihartree deviations for the $2\,^1\Delta$ state of stretched $\ce{CH+}$.
- CIPSI-driven CC(P;Q) with Ndet(in)=1,000 covers <5% of all possible triples but reduces errors to <0.81 millihartree for all states in equilibrium $\ce{CH+}$ and <1.3 millihartree at stretched geometries.
- Increasing Ndet(in) to 5,000 (covering up to 17% of triples) further reduces errors to as low as 0.005–0.207 millihartree, demonstrating systematic convergence toward EOMCCSDT as more triples are included.
- Comparable performance is observed for the CH radical; even strongly multireference states converge to within 0.05–0.37 millihartree relative to EOMCCSDT once 5,000-triplet determinant lists are employed.
Excited State Potential Energy Surfaces (H2O)
The most demanding benchmark involves PES cuts of H2O along an O–H bond stretch for both ground and eleven excited electronic states:
- Standard EOMCCSD and perturbative triples corrections (CR-EOMCC(2,3)) fail for states with strong multireference character and along bond breaking coordinates, yielding non-parallelity errors (NPE) up to 64 millihartree and spurious artifacts in the PES.
- CIPSI-driven CC(P;Q) with Ndet(in)=1,000 substantially ameliorates these failures, with mean unsigned errors (MUE) for all twelve cuts dropping to ≤4.9 millihartree and NPEs ≤14.9 millihartree.
- CC(P;Q)/Ndet(in)=5,000 produces nearly full EOMCCSDT-quality potential curves: MUEs <1.4 millihartree and NPEs <2.6 millihartree for almost all states. Particularly dramatic is the curing of strong artifacts in high-lying multireference excited states, e.g., a 36.8 millihartree excursion in the 3A′′ state is reduced to 1.7 millihartree.
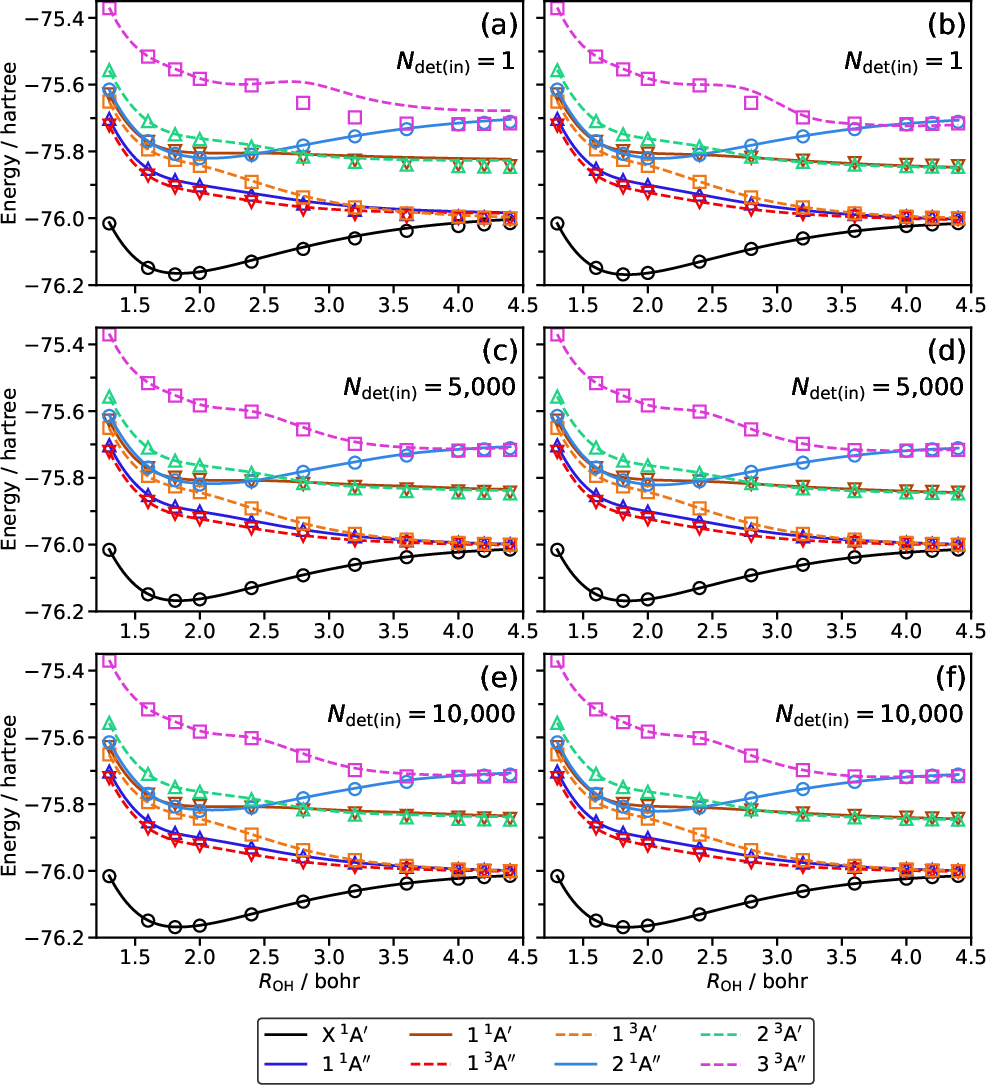
Figure 1: A comparison of the PES cuts of water along the O--H bond breaking coordinate ROH, demonstrating the superior performance of CIPSI-driven CC(P;Q) approaches compared to traditional CCSD(T)/EOMCCSD(T) and CR-EOMCC(2,3), especially for strongly correlated excited states.
These results robustly establish that the CC(P;Q) formalism, when driven by a carefully designed CIPSI determinant selection, achieves near-quantitative accuracy for both ground and electronically excited states at a fraction of the computational cost associated with full CCSDT/EOMCCSDT.
Implications and Future Directions
This extension of the CIPSI-driven CC(P;Q) methodology to excited states has several important ramifications:
- Practical computation of high-level excited-state energies for systems where full triple excitation treatment would otherwise be intractable, including challenging regions such as bond-breaking and strongly correlated excited-state surfaces.
- Systematic, user-controllable convergence from EOMCCSD to EOMCCSDT behavior by simply increasing the CIPSI determinant list, providing a direct diagnostic for correlation recovery and error estimation.
- Efficient resource utilization due to reduction of iterative optimization to a compact (selected) triples space, with non-iterative corrections handled at much lower cost than a single EOMCCSDT cycle.
The method’s performance highlights the deficiency of traditional perturbative and renormalized triples corrections in the presence of strong multireference effects, and offers a compelling substitute that can be tuned for cost versus accuracy.
Future research includes implementing state-specific P spaces using excited-state tailored CIPSI runs, targeting improved convergence for higher excited states. There is also scope for integrating truncated or otherwise adaptive variants of the CIPSI protocol (e.g., selected up to triple or quadruple excitations), which could yield further computational savings.
Conclusion
The extension of the CIPSI-driven CC(P;Q) approach to EOMCC calculations for excited states enables robust and efficient approximation of CCSDT/EOMCCSDT energetics. The formalism achieves high accuracy for vertical, adiabatic, and PES excitations with manageable computational demands, thanks to state-of-the-art selected CI guiding of triple excitation manifold construction. This hybrid, adaptive approach is poised to have substantial impact on the routine application of high-level wave function methods to chemically and spectroscopically crucial excited-state processes, particularly in regimes characterized by strong correlation and multireference character.