Toward Interpretable and Generalizable AI in Regulatory Genomics
Abstract: Deciphering how DNA sequence encodes gene regulation remains a central challenge in biology. Advances in machine learning and functional genomics have enabled sequence-to-function (seq2func) models that predict molecular regulatory readouts directly from DNA sequence. These models are now widely used for variant effect prediction, mechanistic interpretation, and regulatory sequence design. Despite strong performance on held-out genomic regions, their ability to generalize across genetic variation and cellular contexts remains inconsistent. Here we examine how architectural choices, training data, and prediction tasks shape the behavior of seq2func models. We synthesize how interpretability methods and evaluation practices have probed learned cis-regulatory organization and highlighted systematic failure modes, clarifying why strong predictive accuracy can fail to translate into robust regulatory understanding. We argue that progress will require reframing seq2func models as continually refined systems, in which targeted perturbation experiments, systematic evaluation, and iterative model updates are tightly coupled through AI-experiment feedback loops. Under this framework, seq2func models become self-improving tools that progressively deepen their mechanistic grounding and more reliably support biological discovery.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What this paper is about
This paper looks at how AI reads DNA and predicts how genes are turned on or off. Think of DNA as a giant instruction book. Genes are the “pages,” and special short DNA patterns act like switches and dials that control when those pages get read. The authors review today’s AI systems that try to go from DNA letters (A, C, G, T) straight to “how active will this gene be?” They explain what works, what breaks, and how to make these AIs more understandable and reliable—especially when we test DNA changes or switch to different cell types.
The big questions the authors ask
- How do different AI designs, training data, and goals change what these models learn about DNA control?
- Why do models that score well on familiar DNA often struggle on new kinds of DNA changes or in new cell types?
- How can we peek inside these models to see which DNA letters and patterns they rely on—and when those explanations can be trusted?
- What is the right way to test whether a model truly understands cause-and-effect, not just patterns?
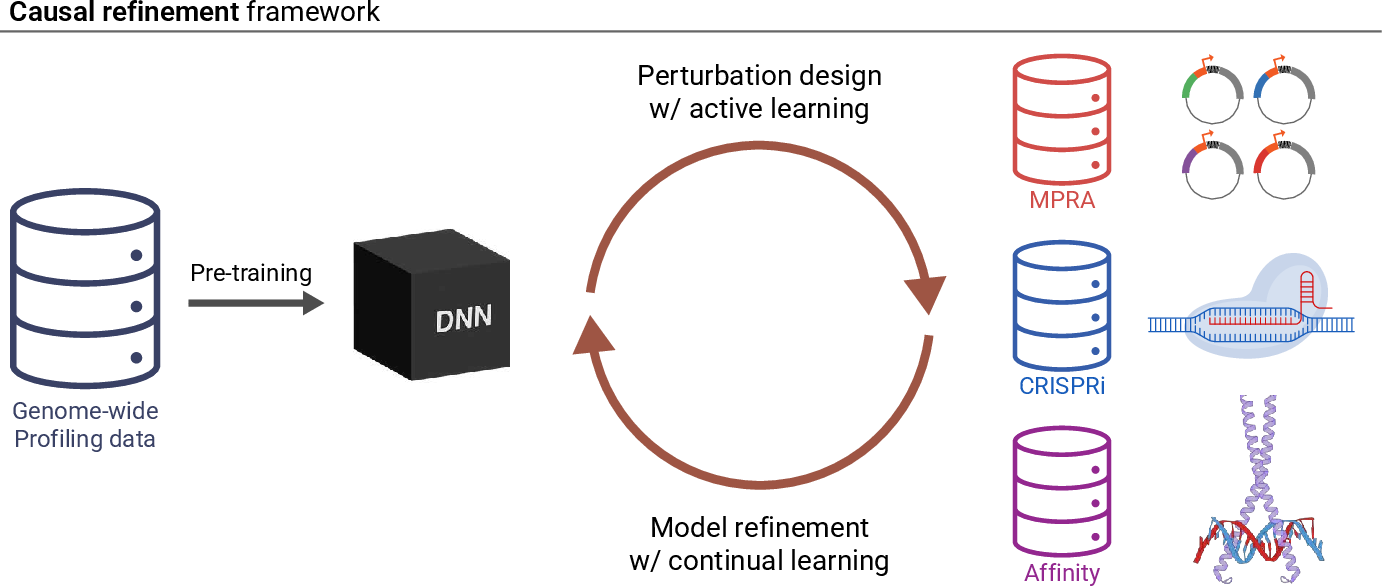
- How can AI and lab experiments work together in a loop so the system keeps getting better over time?
How they study the problem (approach)
This is a review paper, so the authors gather and organize evidence from many recent studies rather than running one new experiment. They explain ideas with simple diagrams and examples, compare model families, and lay out a plan for building better systems. Here are the main pieces of their approach explained in everyday terms:
- Different kinds of DNA-reading AI:
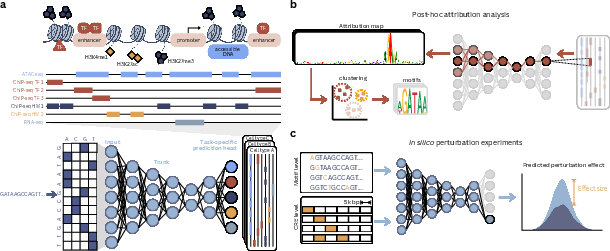
- Seq2func models: These are the main workhorses. They take DNA as input and predict “function” (for example, how open the DNA is, where proteins bind, or how much a gene is used). You can think of them like weather apps that look at today’s map (DNA) and forecast rain (regulatory activity).
- Unsupervised “language” models for genomes: These try to learn patterns from raw DNA without labels, similar to how a phone keyboard guesses the next word. They’re great for proteins, but for regulatory DNA they often miss the important “switch” patterns because most of the DNA background looks similar.
- Generative models: These try to design new DNA that should do a certain job, like writing a new recipe that matches a target flavor.
- Model architecture (the AI’s “shape”):
- Convolutions learn short, local patterns (like recognizing letters and small words—these are called “motifs,” short DNA pieces many proteins recognize).
- Attention and state-space models help combine information across long DNA stretches (like understanding how words form sentences and paragraphs).
- The key idea: gene control works at many scales, so good models stack layers that first find motifs and then learn how those motifs work together.
- Data and tasks (what we teach the AI to do):
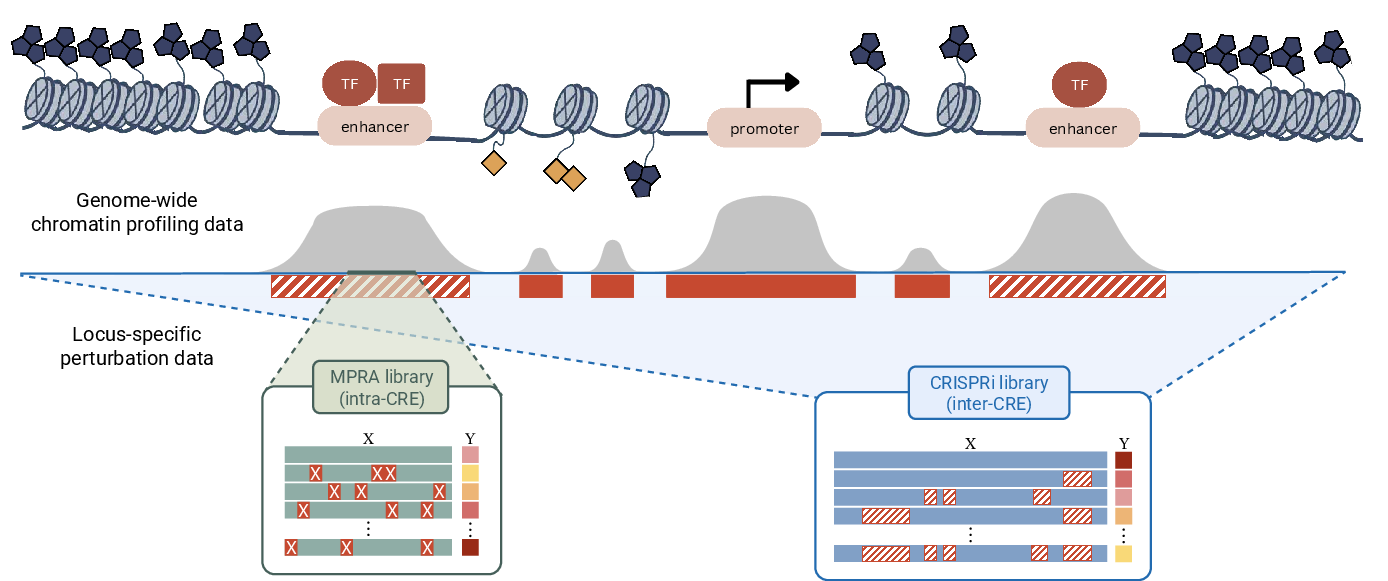
- Genome-wide assays: measurements across the genome in real cells (for example, where certain proteins bind DNA).
- MPRAs (massively parallel reporter assays): lab tests that measure activity of thousands of short sequences, often with many tiny changes, to see what boosts or lowers activity.
- Training models to predict detailed, quantitative signals (not just yes/no) usually teaches them richer, more realistic rules.
- Looking inside the models (interpretability):
- Attribution maps: highlight which DNA letters matter most for a prediction—like underlining important words in a sentence.
- Virtual perturbations (“in silico experiments”): change a letter, add/remove a motif, move things around in a simulated way to see how the model’s prediction changes. This tests cause and effect according to the model.
- Clustering many explanations can reveal common motifs and how they interact.
- Testing generalization (do they work on new situations?):
- The authors describe three kinds of “shifts” that break models:
- New inputs: the DNA looks different (for example, new mutations or synthetic sequences).
- New rulers: the lab measurement changes (different assay tools can read the same biology differently).
- New rules: the biology itself changes (a different cell type or state, so the same DNA acts differently).
- Many current tests are too easy and don’t catch these problems.
- A plan for self-improving AI:
- Use an “AI ↔ experiment” loop: AI makes predictions and suggests informative DNA changes; labs test them; results feed back to update the AI.
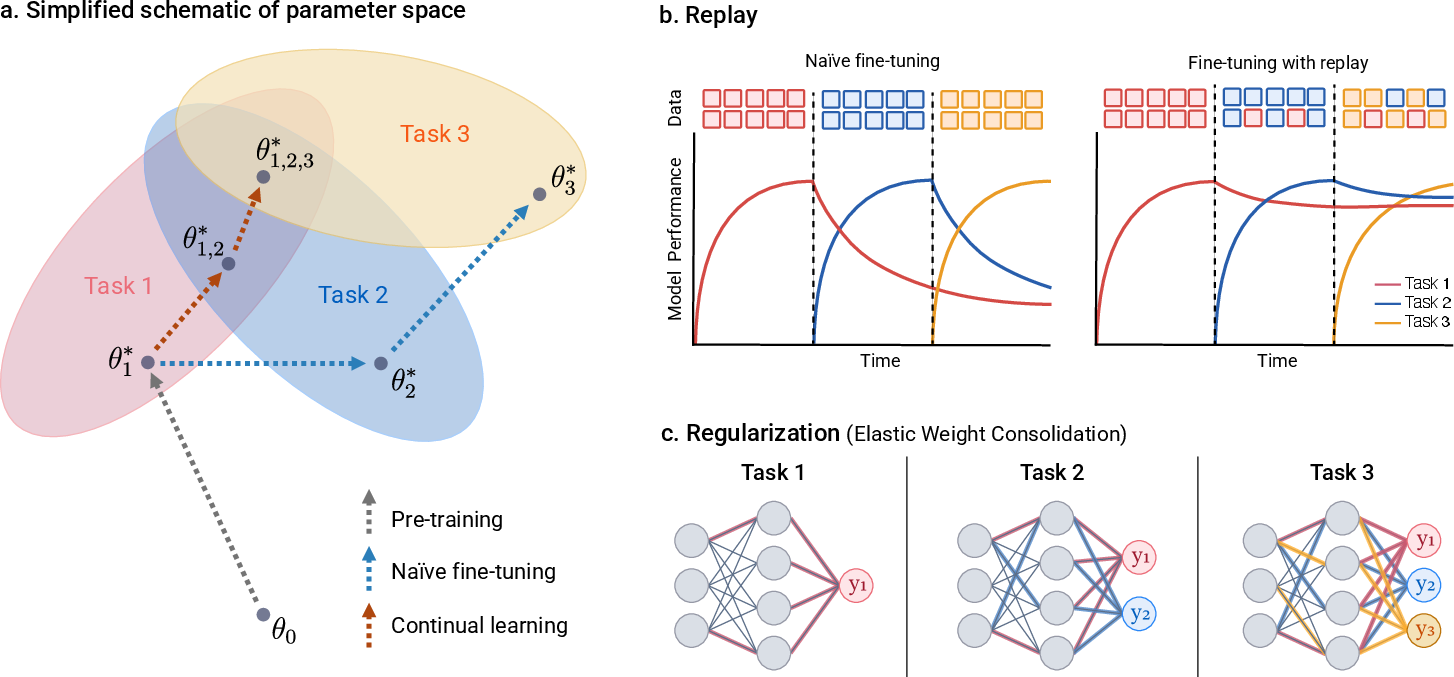
- Use continual learning: update the model with new data without forgetting what it already learned.
- Design smarter perturbations (not just random tweaks) and use active learning to pick the most informative experiments.
What they learned (main takeaways)
- Strong scores don’t guarantee real understanding. Many models are excellent at predicting familiar patterns seen during training but stumble when facing new kinds of DNA changes, long-range interactions, or different cell types.
- Architecture matters, but data matters more. Sophisticated models help, yet what you ask them to predict and which data you provide largely determine what rules they learn.
- Interpreting models is useful but tricky. Heatmaps and motif finders can reveal important patterns, but they can be misleading if the model itself isn’t reliable in that situation. It’s important to confirm ideas with virtual (and, when possible, real) perturbations.
- Current tests often miss hidden weaknesses. Simple train/test splits can let similar sequences leak across sets, making performance look better than it truly is. We need tougher, more realistic benchmarks, especially those involving perturbations.
- The biggest gap is causal knowledge. Training only on “what nature already did” (the reference genome) covers a very tiny part of possible DNA. To learn deeper cause-and-effect rules, models need dense, targeted perturbation data that show how changing sequence changes function.
- The solution is a feedback loop. Combine broad genome-wide data (big-picture coverage) with focused perturbation assays (high-detail, causal insights), and continually update the model to preserve old skills while learning new ones.
- Smarter experiment design helps everyone. Thoughtful, diversity-rich, yet biologically realistic DNA edits—and active learning to choose them—can speed up discovery and make models more general and trustworthy.
Why this matters and what could happen next
If AI can reliably read and design the “switches” in DNA, scientists could:
- Better predict which genetic changes might cause disease.
- Design safer, more effective gene switches for therapies.
- Understand how the same DNA behaves differently across cell types, development, and disease.
- Build trustworthy, testable, and explainable tools for biology, not just black boxes.
The authors’ main message is practical: treat these DNA-reading AIs like growing students. Give them the right lessons (richer data), ask the right questions (tough tests), check their work (interpretability and perturbations), and keep teaching them over time (continual learning). With this loop of AI-guided experiments and experiment-guided AI, models can become more interpretable, more general, and more useful for real biological discovery.
Knowledge Gaps
Unresolved Knowledge Gaps, Limitations, and Open Questions
Below is a consolidated list of concrete gaps and open questions that remain unresolved and could guide future research.
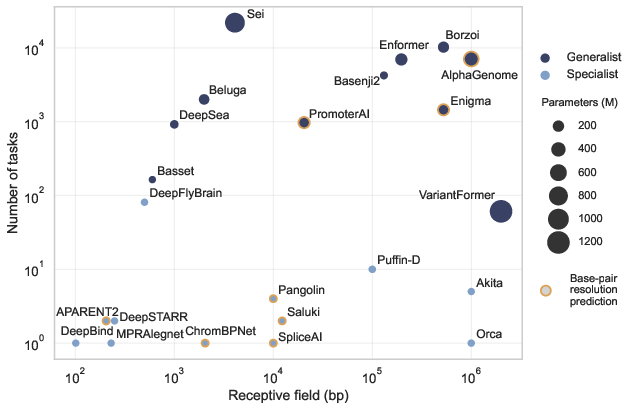
- Lack of standardized, fair comparisons of architectural inductive biases: controlled protocols to compare CNNs, transformers, and state-space models under identical tuning, data budgets, and receptive fields, with metrics for data efficiency, robustness to hyperparameters, and sensitivity to optimization choices.
- Insufficient evidence that unsupervised genomic LLMs (gLMs), especially those restricted to regulatory regions or augmented with conservation, learn actionable cis-regulatory logic; need benchmarks and probes that directly test mechanistic understanding versus statistical association.
- Discrete-diffusion generative methods remain small-scale; unclear if they recover regulatory grammar at genome scale and under multi-cell-type contexts—scaling strategies and rigorous evaluation suites are needed.
- Persistent multitask bias toward shared housekeeping programs: general methods to prevent loss domination by shared tasks and to amplify context-specific signals (beyond ad hoc upsampling, focal losses, or fine-tuning) remain unidentified; require metrics stratified by differentially regulated elements.
- Ambiguity in profile attribution reduction: standardized practices for deriving scalar outputs from profile predictions (or broader adoption of position-wise methods like PISA), and quantitative tests for attribution stability and fidelity across noise regimes.
- Gradient-based attribution reliability under discrete sequence manifolds: when and how statistical gradient correction and gauge-fixing yield faithful importance scores across different architectures, assays, and sequence contexts; ground-truth validation against biophysical measurements.
- Robust disentangling of additive versus interaction effects: scalable frameworks (e.g., gauge fixing, DIAMOND, second-order ISM/Hessians) to separate true interactions from apparent non-additivity, with genome-wide validation and controls for confounders.
- Attention-derived motif–motif interaction maps (e.g., GLIFAC) lack systematic ground-truth validation; need experiments to verify inferred interactions and quantify false discovery rates across models and assays.
- Sparse autoencoder (SAE) latents are often fragmented or absorbed; methods to reduce feature splitting/merging, criteria to confirm mechanistic meaning, and standardized virtual perturbation protocols to validate latent features are missing.
- Community benchmarks for generalization under covariate, label, and concept shifts are incomplete: require perturbation-rich, multi-assay, multi-cell-type suites with biologically interpretable metrics, success criteria, and longitudinal stability akin to ImageNet/CASP.
- Dataset splitting protocols do not reliably prevent information leakage via compositional similarity; need sequence-similarity-aware splits and diagnostic tools to detect leakage across chromosomes and related regions.
- Measurement-process modeling is underdeveloped: general frameworks to disentangle latent biology from assay-specific biases/noise (label shift), and standardized procedures to quantify and correct distortions across platforms.
- Unclear utility of cross-species diversity for human regulatory generalization: when and how interspecies variation improves mechanism learning versus introduces confounding; require prospective, cross-species perturbational validations.
- Bridging resolution–coverage tradeoff remains unsolved: principled strategies to fuse genome-wide profiling (broad, low resolution) with locus-specific perturbations (narrow, high resolution) to learn transferable mechanisms that generalize genome-wide.
- Genomics-specific continual learning is unproven: which combinations of replay, regularization, architecture partitioning, or nested learning best integrate diverse perturbation datasets while avoiding catastrophic forgetting; metrics for forward and backward transfer tailored to regulatory tasks.
- Replay content and strategy are unclear: what to store or synthesize (e.g., representative loci, perturbational “mechanism exemplars,” or generative replay) to preserve broad capabilities while incorporating new mechanistic constraints.
- Active learning remains weakly coupled to information gain: acquisition functions need to move beyond uncertainty/diversity heuristics to mechanism-aware criteria; joint design of candidate reservoirs and acquisition rules under experimental constraints is largely unexplored.
- Evolution-inspired perturbations (e.g., inversions, translocations, EvoAug-style edits) lack systematic experimental evaluation in mammalian systems; need to identify perturbation primitives that maximize mechanistic yield and dynamic-range balance.
- Closed-loop AI–experiment systems lack validated pipelines and benchmarks: protocols for hypothesis generation, sequence design, acquisition, measurement, and model updating; safeguards against assay-specific overfitting, drift, and confirmation bias; stopping criteria and governance.
- External validation of virtual assays is limited: prospective, multi-locus campaigns to test necessity/sufficiency, positional/spacing rules, and combinatorial grammar claims across cell types and assays; standardized reporting for effect-size calibration.
- Modeling cellular context (concept shift) is incomplete: how to represent dynamic trans-factor concentrations, chromatin state, signaling, and developmental trajectories in seq2func models; strategies to transfer across cell types/states without retraining.
- Scaling laws for foundation-style genomic models are unknown: how capacity, input length, task breadth, and data diversity translate into generalization and mechanistic fidelity; identification of diminishing returns and bottlenecks.
- Quantitative metrics for inductive bias are missing: measures that relate learned dependencies to receptive-field structure, locality versus long-range integration, and hierarchical organization across layers.
- Uncertainty calibration under distribution shift is underdeveloped: methods to produce well-calibrated uncertainty for variant effects, synthetic designs, and structural rearrangements; evaluation protocols that link uncertainty to experimental validation rates.
- Handling extreme covariate shifts (de novo mammalian sequences with low activity): strategies to design libraries with balanced dynamic ranges (e.g., scaffolded sequences, motif seeding), and to avoid trivial negatives dominating training or evaluation.
- Reproducibility and reporting standards need strengthening: disentangling tuning effort from architectural gains, cross-lab reproducibility for preprocessing and evaluation, and minimal documentation for splits, metrics, and statistical testing.
- Automated interpretability remains aspirational: validation frameworks for agentic systems and LLM-guided surrogate modeling to ensure correctness, reduce hallucinations, and quantify hypothesis hit rates and mechanistic precision.
- Encoding mechanistic priors into architectures is ad hoc: which biophysical constraints (e.g., occupancy models, cooperative binding, symmetry) improve generalization without overfitting, and how to formalize/justify them theoretically and empirically.
Practical Applications
Immediate Applications
Below are practical use cases that can be deployed now, derived from the paper’s findings on seq2func modeling, interpretability, evaluation, and perturbation-driven analysis.
- Variant effect prioritization for noncoding regions (Healthcare, Pharma)
- Use profile-trained seq2func models (e.g., ChromBPNet, Enformer/Borzoi/AlphaGenome family) to score SNVs and small indels affecting chromatin accessibility, TF binding, and transcription initiation; triage variants for follow-up functional validation in rare disease and oncology pipelines.
- Tools/workflows: virtual assays on patient-specific loci; in silico mutagenesis (ISM), PISA for base-pair–aligned attribution; global importance analysis for causal effect sizing.
- Assumptions/dependencies: assay-aware calibration to mitigate label shift; variant interpretation standards; access to matched functional genomics data; cautious use beyond closely related training contexts.
- Hypothesis generation and mechanistic mapping of cis-regulatory grammar (Academia, Biotech R&D)
- Apply complementary interpretability methods to uncover motifs, syntax, and interactions: TF-MoDISco for motif discovery, DFIM/Hessian-based probes for pairwise effects, gauge-fixing to separate additive vs interaction components; use SEAM and SQUID to summarize dense perturbation libraries into interpretable rules.
- Tools/workflows: standardized “interpretation stack” combining attribution + virtual perturbation + surrogate modeling; attention aggregation (GLIFAC) for motif–motif co-occurrence.
- Assumptions/dependencies: model robustness under distribution shift; multi-assay, multi-cell-type evaluation to confirm generality; tuning for noise and gradient correction.
- Assay bias correction and improved profiling readouts (Genomics Core Labs, Software)
- Incorporate measurement-process modeling and bias correction (e.g., Tn5 bias in ATAC-seq) to improve signal interpretation and peak calling; profile regression models reduce false positives/negatives.
- Tools/workflows: assay-aware training, bias correction modules (e.g., ChromBPNet-style); QC dashboards with distribution-shift diagnostics.
- Assumptions/dependencies: availability of control datasets; reproducible pipelines; validation against orthogonal assays.
- Targeted CRISPR perturbation design and MPRA library optimization (Academia, Biotech R&D)
- Use virtual perturbation campaigns to prioritize guide targets, motif edits, and element rearrangements likely to yield interpretable, high-dynamic-range readouts; design motif-centric and partial-random libraries.
- Tools/workflows: eugene and Tangermeme for programmable motif insertions; batch-mode selection of sequences informed by global importance analysis.
- Assumptions/dependencies: model precision at loci of interest; lab capacity for pooled screens; mitigation of acquisition bias toward uninformative extremes.
- Yeast and simple-system synthetic promoter/enhancer design (Synthetic Biology)
- Design functional regulatory sequences in organisms where random or lightly structured designs can drive expression; deploy for pathway balancing, biosensors, and circuit prototyping.
- Tools/workflows: sequence-design models guided by virtual assays; evolutionary augmentations (EvoAug) for robust training; rapid screening via episomal MPRAs.
- Assumptions/dependencies: species/context specificity; limited transferability to mammalian systems without additional constraints.
- Benchmarking and internal auditing under realistic distribution shifts (Academia, Software/AI)
- Adopt stratified evaluation that probes covariate, label, and concept shift; calibrate performance on progressively out-of-distribution perturbations; use community efforts (e.g., GAME) where available.
- Tools/workflows: curated benchmark suites with locus-level and combinatorial edits; leakage detection in splits; reporting protocols tied to biologically interpretable metrics.
- Assumptions/dependencies: access to perturbational datasets; organizational commitment to rigorous evaluation beyond random/chromosome splits.
- Education and training aids for regulatory genomics (Education)
- Use attribution maps and perturbation simulations in teaching labs to demonstrate motif function, sufficiency/necessity tests, and combinatorial logic.
- Tools/workflows: interactive notebooks bundling TF-MoDISco, PISA, GI; small-scale synthetic-locus MPRA design exercises.
- Assumptions/dependencies: open-source models and datasets; basic sequencing/assay access or simulated data.
Long-Term Applications
The following applications depend on further research, scaling, new datasets, or deeper model generalization; they align with the paper’s vision of AI–experiment feedback loops, continual learning, and standardized evaluation.
- Clinically validated decision support for noncoding variant pathogenicity (Healthcare, Policy)
- Integrate seq2func predictions with clinical genomics to classify regulatory variants impacting disease; establish regulatory-grade pipelines with prospective validation and standardized thresholds.
- Tools/products: clinical-grade “Regulatory Variant Interpreter” with assay-aware calibration, uncertainty quantification, and audit trails.
- Assumptions/dependencies: robust cross-cell-type generalization (concept shift); regulatory approval; multi-center trials; harmonized data standards.
- AI–experiment feedback loops for self-improving regulatory models (Biotech R&D, Academia, Software/AI)
- Build closed-loop platforms that propose, execute, and learn from perturbation assays (MPRA, CRISPRi), continually refining models without catastrophic forgetting; leverage replay/regularization and task-aware modules.
- Tools/products: “Regulatory Model OS” integrating active learning, lab automation, reservoir generation (Genome-Shuffle-seq, generative reservoirs), continual learning schedulers.
- Assumptions/dependencies: scalable synthesis and screening; standardized metadata; compute for iterative training; robust acquisition functions aligned to information gain.
- Foundation-style regulatory genomics models across species and assays (Biotech R&D, Agriculture, Pharma)
- Train multitask, long-context models that generalize across assays, cell types, and species; serve as virtual assays with base-pair resolution and interpretable heads for TFs and chromatin features.
- Tools/products: cross-species “Regulatory Foundation Model” with assay-aware adapters; motif-aware heads; API for virtual profiling and design.
- Assumptions/dependencies: large, balanced multi-assay datasets; rigorous benchmarks spanning covariate/label/concept shifts; attention to inductive biases for data efficiency.
- Safe and reliable de novo mammalian regulatory sequence design (Gene Therapy, Synthetic Biology)
- Generate and validate tissue-specific, context-stable promoters/enhancers for AAV vectors, CAR-T constructs, and cell therapies; incorporate biophysical constraints and safety checks into generative objectives.
- Tools/products: design suite combining conditional generative models (diffusion/gLMs) with assay-aware scoring, off-target risk assessment, and in vivo validation workflows.
- Assumptions/dependencies: accurate modeling of context dependence; regulatory safety guidelines; comprehensive off-target and immunogenicity screens.
- LLM-guided mechanistic discovery and global surrogate modeling (Academia, Software/AI)
- Deploy agentic AI systems to orchestrate interpretation stacks, propose structured surrogate models, and run program-search for mechanistic rules (e.g., AlphaEvolve, LLMGEN); automate hypothesis generation and experiment planning.
- Tools/products: “Agent Scientist” for cis-regulatory grammar discovery with integrated knowledge bases and experimental design modules.
- Assumptions/dependencies: reliable tool-use and verification; human-in-the-loop oversight; guardrails against spurious correlations; standardized reporting of uncertainty.
- Assay harmonization and latent measurement-process modeling (Genomics Core Labs, Policy)
- Explicitly model measurement processes to disentangle latent regulatory activity from assay-specific distortions; enable cross-platform comparability and meta-analysis.
- Tools/products: assay adapters and latent-variable “regulatory decoders” that align MPRA, CRISPR screens, ATAC-seq, ChIP-seq, and QTL signals.
- Assumptions/dependencies: detailed assay metadata; calibration datasets; acceptance of harmonization standards by the community.
- Active-learning–driven national perturbation programs (Academia, Biotech R&D, Policy)
- Coordinate multi-institutional efforts to generate dense, structured perturbation datasets across representative loci, cell types, and biological scales; benchmark information gain and transfer.
- Tools/products: “MPRA Studio” + “CRISPRi Atlas” with shared reservoirs, acquisition strategies (batch-mode), and FAIR data publishing.
- Assumptions/dependencies: funding and infrastructure; consensus on acquisition metrics; lab automation; ethical/data-sharing frameworks.
- Crop trait engineering via regulatory rewiring (Agriculture)
- Design tissue- and condition-specific plant promoters/enhancers to optimize yield, stress tolerance, and nutrient content; transfer regulatory rules across cultivars/species.
- Tools/products: plant-specific foundation models; generative design with field-ready validation pipelines.
- Assumptions/dependencies: plant transformation efficiency; species-specific datasets; environmental robustness; regulatory approval for GM crops.
- Standards and safety frameworks for AI-designed sequences (Policy, Biosecurity)
- Establish governance for risk assessment, auditability, and provenance of AI-generated regulatory sequences; define acceptable performance under distribution shifts and reporting of uncertainties.
- Tools/products: certification protocols, conformance test suites, sequence provenance ledgers.
- Assumptions/dependencies: stakeholder consensus; international coordination; integration with biosafety regulations.
- Community benchmarks and competitions for regulatory generalization (Academia, Software/AI)
- Launch CASP/ImageNet-style long-running benchmarks emphasizing perturbational evaluation across shifts; track cumulative progress and enable fair comparisons.
- Tools/products: publicly curated datasets, standardized metrics tied to biological outcomes, leaderboards with interpretability and generalization scoring.
- Assumptions/dependencies: sustained funding; broad participation; transparent governance and data stewardship.
- Public-facing educational simulators for genomics (Education, Daily life)
- Create interactive apps that let users explore how DNA sequence controls gene regulation via virtual assays; demystify genome function for students and citizen scientists.
- Tools/products: web-based simulators with safe toy models; curriculum-aligned modules.
- Assumptions/dependencies: simplified, explainable models; responsible communication of uncertainty and limitations.
Glossary
- Active learning: A strategy that selects the most informative experiments or data points to query next, often based on model uncertainty or expected information gain. "Active learning offers a complementary, principled framework by prioritizing perturbations expected to yield maximal information gain"
- Agentic AI systems: AI tools that autonomously plan and coordinate sequences of analyses or experiments to pursue goals, such as mechanistic discovery. "One is agentic AI systems that orchestrate existing interpretability tools to identify recurring regulatory patterns, synthesize results, generate mechanistic hypotheses, and propose targeted virtual perturbations."
- ATAC-seq: A functional genomics assay that profiles open chromatin using Tn5 transposase insertion. "Tn5 insertion bias in ATAC-seq"
- Backward transfer: In continual learning, improvement on earlier tasks after training on new tasks. "while refinement on new loci can sharpen previously learned mechanisms (backward transfer)"
- Batch-mode active learning: An active learning setup that selects informative batches of sequences jointly rather than one at a time. "In regulatory genomics, this problem is naturally posed as batch-mode active learning"
- Biophysical measurements: Quantitative experimental assays that characterize physical interactions or energies (e.g., TF–DNA binding energetics). "learn motif representations whose quantitative preferences align with biophysical measurements"
- Catastrophic forgetting: The loss of previously learned knowledge when a model is fine-tuned on new data. "risks catastrophic forgetting"
- Chromatin accessibility: The openness of chromatin that permits regulatory factor binding, often measured by assays like ATAC-seq or DNase-seq. "including chromatin accessibility, TF binding, histone modifications, transcription initiation, and steady-state gene expression"
- Chromothripsis: A massive genomic rearrangement process involving many breakages and reassemblies in a single event. "analogous to chromothripsis"
- Cis-regulatory code: The set of principles by which DNA sequence encodes regulatory activity in specific cellular contexts. "cis-regulatory code: the principles by which DNA sequence specifies regulatory activity in a given cellular context."
- Cis-regulatory element (CRE): A DNA region (e.g., enhancer, promoter) that regulates gene expression of nearby genes. "Massively parallel reporter assays (MPRAs) introduce dense sequence variation within cis-regulatory elements (CREs) to measure how local nucleotide changes affect regulatory activity."
- Concept shift: A distribution shift where the underlying mapping from inputs to outputs changes (e.g., across cell types). "Concept shift represents a more severe regime in which the sequence–function mapping itself changes."
- Conditional generative models: Generative models that produce sequences conditioned on specified functional or contextual variables. "Conditional generative models learn sequence distributions explicitly conditioned on functional or contextual variables"
- Covariate shift: A distribution shift where the input distribution changes but the input–output mapping remains the same. "Covariate shift occurs when the input sequence distribution changes while the underlying sequence–function mapping remains fixed."
- CRISPR interference (CRISPRi): A CRISPR-based technique to repress gene or element activity without cutting DNA, often to test necessity. "CRISPR interference (CRISPRi) libraries target entire CREs, individually or in combination, within their native chromatin context"
- CRISPR-based screens: Pooled perturbation experiments using CRISPR to systematically perturb genes or regulatory elements and measure effects. "CRISPR-based screens"
- DFIM: Deep Feature Interaction Maps; a method combining perturbation and attribution to quantify feature interactions. "DFIM"
- DIAMOND: A statistical testing framework to disambiguate interaction effects from non-additivity in model interpretations. "DIAMOND"
- Dilated convolutions: Convolutional layers with gaps that expand receptive fields without increasing parameter count, enabling long-range context. "Dilated convolutions and pooling layers accelerate this expansion while preserving locality consistent with motif syntax."
- Discrete-diffusion approaches: Generative modeling methods over discrete sequences that simulate mutation-like transitions toward functional sequences. "Discrete-diffusion approaches offer a promising alternative by modeling mutational transitions that evolve random sequences toward functional ones"
- Elastic weight consolidation: A continual learning regularization that constrains updates to parameters important for previous tasks. "Regularization-based methods, such as elastic weight consolidation, penalize updates to parameters estimated to be important for earlier tasks"
- EvoAug: Evolution-inspired data augmentations that apply realistic sequence edits to improve generalization. "evolution-inspired data augmentations via EvoAug have proven effective in silico"
- Expression QTLs: Genetic variants associated with differences in gene expression levels across individuals or tissues. "expression QTLs"
- Flat minima: Regions of the loss landscape where many parameter configurations yield similar low loss, often linked to robustness. "often residing in relatively flat regions of the loss landscape"
- Forward transfer: In continual learning, improvement on future tasks thanks to knowledge from earlier tasks. "Insight gained at one locus can improve predictions at others with similar regulatory architectures (forward transfer)"
- Functional genomics: High-throughput experimental approaches that measure molecular activities (e.g., binding, accessibility, expression) across the genome. "Advances in machine learning and functional genomics have enabled sequence-to-function (seq2func) models"
- Gauge-fixing: A procedure to separate additive and interaction components in interpretations without changing the underlying function. "gauge-fixing approaches that separate additive and interaction components without altering the underlying function"
- Genomic LLMs (gLMs): Large sequence models trained self-supervised on genomic sequences to learn representations from sequence statistics. "genomic LLMs (gLMs)"
- Genome-Shuffle-seq: An experimental strategy that creates structured, evolution-inspired genomic rearrangements to probe regulatory logic. "Genome-Shuffle-seq achieves this through deletions, inversions, translocations, and mutations."
- Genome-wide profiling assays: Experiments that measure regulatory activity across the genome in native context (e.g., ChIP-seq, ATAC-seq). "genome-wide profiling assays"
- GLIFAC: An aggregation framework that summarizes attention-weighted motif co-occurrences to infer global motif–motif interactions. "GLIFAC summarize attention-weighted motif co-occurrences across many sequences"
- Global importance analysis: A marginalization-based perturbation framework to estimate the causal effect of features on model predictions. "This marginalization strategy, formalized in global importance analysis, isolates the causal effect of the perturbed pattern through the lens of the model."
- Global surrogate modeling: Fitting interpretable, global functions that approximate complex model behavior to extract mechanistic rules. "LLM-guided global surrogate modeling, in which interpretable functions are discovered and refined through closed-loop program-search procedures"
- Hessians: Second-order derivatives that capture curvature of the loss or interaction effects between input features. "second-order gradient methods like Hessians"
- Housekeeping regulatory programs: Regulatory features common across many cell types that drive shared basal cellular functions. "such as housekeeping regulatory programs"
- In silico mutagenesis (ISM): A perturbation technique that individually mutates positions in a sequence to assess their effect on model predictions. "in silico mutagenesis (ISM)"
- Label shift: A distribution shift where measurement processes change while the underlying biology is the same, altering observed labels. "Label shift arises when the input distribution is similar, but different assays measure the same underlying biological activity and produce systematically different observed measurements."
- Loss landscape: The surface of the loss function with respect to model parameters, shaping optimization dynamics and solutions. "architecture and training data jointly define the loss landscape"
- Motif syntax: Organizational rules (spacing, orientation, combinations) governing how sequence motifs interact to drive regulation. "preserving locality consistent with motif syntax"
- PISA: A profiling interpretation method that provides position-specific attribution maps from profile predictions. "PISA avoids this ambiguity by providing attribution maps at individual positions of the predicted profile"
- Replay-based methods: Continual learning techniques that interleave past data with new data to reduce forgetting. "Replay-based methods maintain performance over time by mixing past examples with new data during fine-tuning."
- Reverse-complement weight sharing: An architectural constraint tying parameters for sequences and their reverse complements to enforce DNA symmetry. "reverse-complement weight sharing reduce redundancy"
- Saturation mutagenesis: Comprehensive single-nucleotide substitution scanning to map functional sensitivity across a sequence. "Saturation mutagenesis introduces single-nucleotide changes and is widely used for its tractability"
- SEAM: A method that clusters full attribution maps from dense mutagenesis to reveal accessible regulatory mechanisms. "SEAM instead clusters full attribution maps derived from dense mutagenesis libraries of a reference sequence"
- Self-attention: A mechanism that models pairwise interactions across all positions in a sequence, enabling long-range dependencies. "Transformers use multi-head self-attention to capture pairwise interactions across the full sequence"
- Sloppy modes: Weakly constrained directions in parameter space that allow adaptation without degrading prior performance. "sloppy modes"
- Sparse autoencoders (SAEs): Models that decompose dense representations into sparse latent factors intended to capture interpretable features. "Sparse autoencoders (SAEs) decompose uninterpretable dense embeddings into sparse latent features"
- Squeeze–excitation: Modules that adaptively reweight channels based on global context to emphasize informative features. "Squeezeâexcitation modules provide context-dependent channel reweighting"
- State-space models: Sequence models that propagate information with structured recurrent or gating dynamics scaling linearly with sequence length. "state-space models"
- Surrogate modeling: Building simpler, interpretable models to approximate and explain predictions of complex seq2func models. "surrogate modeling approaches that fit structured, interpretable functions to approximate seq2func predictions."
- TF-MoDISco: A clustering method that aggregates high-attribution subsequences into motif-like patterns for global interpretation. "TF-MoDISco clusters recurrent high-attribution subsequences into motif-like patterns"
- Tn5 insertion bias: Sequence preferences of the Tn5 transposase that introduce technical bias in chromatin accessibility assays. "Tn5 insertion bias in ATAC-seq"
- Transcription factor (TF): A protein that binds specific DNA motifs to regulate gene expression. "transcription factor (TF) binding"
- Virtual perturbation experiments: In silico tests (insertions, deletions, rearrangements) that probe causal effects of sequence features using the model as a surrogate assay. "virtual perturbation experiments can provide a more direct test of whether SAE latents correspond to specific regulatory mechanisms."
Collections
Sign up for free to add this paper to one or more collections.