Scalable Spatio-Temporal SE(3) Diffusion for Long-Horizon Protein Dynamics
Abstract: Molecular dynamics (MD) simulations remain the gold standard for studying protein dynamics, but their computational cost limits access to biologically relevant timescales. Recent generative models have shown promise in accelerating simulations, yet they struggle with long-horizon generation due to architectural constraints, error accumulation, and inadequate modeling of spatio-temporal dynamics. We present STAR-MD (Spatio-Temporal Autoregressive Rollout for Molecular Dynamics), a scalable SE(3)-equivariant diffusion model that generates physically plausible protein trajectories over microsecond timescales. Our key innovation is a causal diffusion transformer with joint spatio-temporal attention that efficiently captures complex space-time dependencies while avoiding the memory bottlenecks of existing methods. On the standard ATLAS benchmark, STAR-MD achieves state-of-the-art performance across all metrics--substantially improving conformational coverage, structural validity, and dynamic fidelity compared to previous methods. STAR-MD successfully extrapolates to generate stable microsecond-scale trajectories where baseline methods fail catastrophically, maintaining high structural quality throughout the extended rollout. Our comprehensive evaluation reveals severe limitations in current models for long-horizon generation, while demonstrating that STAR-MD's joint spatio-temporal modeling enables robust dynamics simulation at biologically relevant timescales, paving the way for accelerated exploration of protein function.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What is this paper about?
Proteins aren’t still statues—they wiggle, bend, and change shape over time to do their jobs in our bodies. Scientists usually study these motions using super-detailed physics simulations called molecular dynamics (MD). MD is very accurate but painfully slow, often taking days or weeks to simulate just tiny slices of time.
This paper introduces STAR-MD, an AI model that makes realistic “movies” of protein motion much faster. It’s designed to create long, stable, and physically sensible protein trajectories (think: a smooth animation of a protein moving) that can stretch to important biological timescales—up to microseconds—where many interesting changes actually happen.
What questions were the researchers trying to answer?
The authors focused on three simple questions:
- Can we build an AI that makes long, realistic protein movies, not just short clips?
- Can the model remember what happened earlier so it doesn’t drift off into nonsense over time?
- Can it work efficiently on bigger proteins without running out of memory?
How does the method work? (Explained with everyday ideas)
Think of making a flipbook animation:
- Each page is a frame of the protein’s shape.
- To draw the next page, you look at previous pages and keep things consistent so the motion looks natural.
STAR-MD does something similar, one frame at a time.
Here are the key ideas, with simple analogies:
- Autoregressive generation: The model makes the animation one frame at a time, always looking back at everything it has already drawn. Like writing a story sentence by sentence, making sure the plot stays consistent.
- Diffusion model: To draw each frame, the model starts with a “noisy” or messy version of the frame and then cleans it up step by step. It’s like un-blurring a noisy photo until the picture becomes clear.
- Physics-aware geometry (SE(3)-equivariance): Proteins can be rotated or shifted in space without changing what they are. The model is built to respect this—if you rotate the protein, the model’s predictions rotate in the same way. This helps keep the physics realistic.
- Joint spatio-temporal attention: “Attention” is a way for the model to focus on what matters. STAR-MD looks at space (which parts of the protein) and time (which past frames) at the same time, not separately. Imagine tracking a dance move: you don’t just watch one dancer or one moment—you watch how different dancers move together across time.
- Training to be stable over long runs: The team adds a tiny bit of noise to past frames during training—and does the same thing when generating new frames. This teaches the model to handle small imperfections and prevents small mistakes from snowballing into big errors.
- Flexible timing: The model learns with many different time gaps between frames (like 1 ns, 2 ns, etc.). That way, later it can make animations at different speeds without retraining.
In short, STAR-MD is a smart, physics-aware animator that builds a protein movie frame by frame, using past frames wisely and efficiently.
What did they find, and why does it matter?
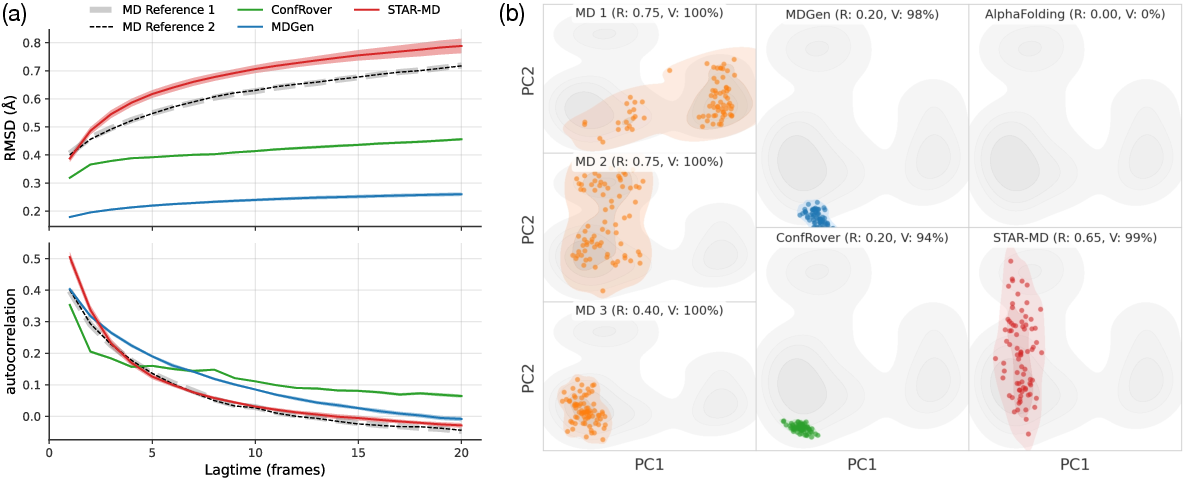
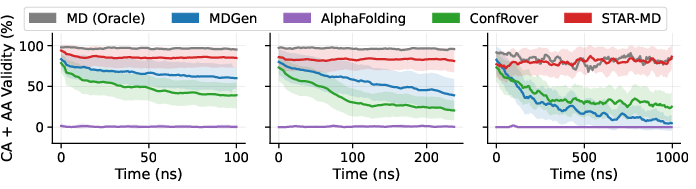
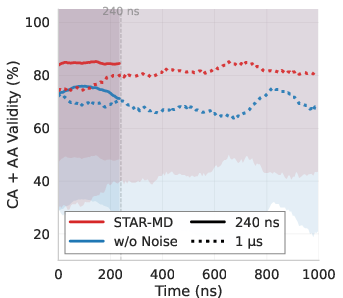
The researchers tested STAR-MD on a large benchmark called ATLAS, which contains many 100-nanosecond protein simulations, and then pushed further to 240 nanoseconds and even 1 microsecond. They compared to strong previous AI methods.
Here’s what they found:
- More realistic structures: STAR-MD produced far more physically valid protein shapes than other models (around the ~85–87% range on standard tests), while some baselines produced many broken or unrealistic structures.
- Better coverage of real motions: It explored more of the real movements seen in reference MD simulations, meaning its “movies” look more like what nature (and MD) shows.
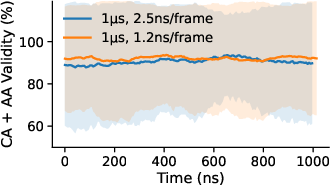
- Stable over long times: Unlike other methods that fall apart as the animation gets longer, STAR-MD stayed stable and realistic even out to a microsecond—where many biologically important events happen.
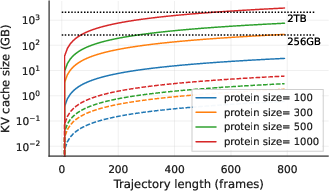
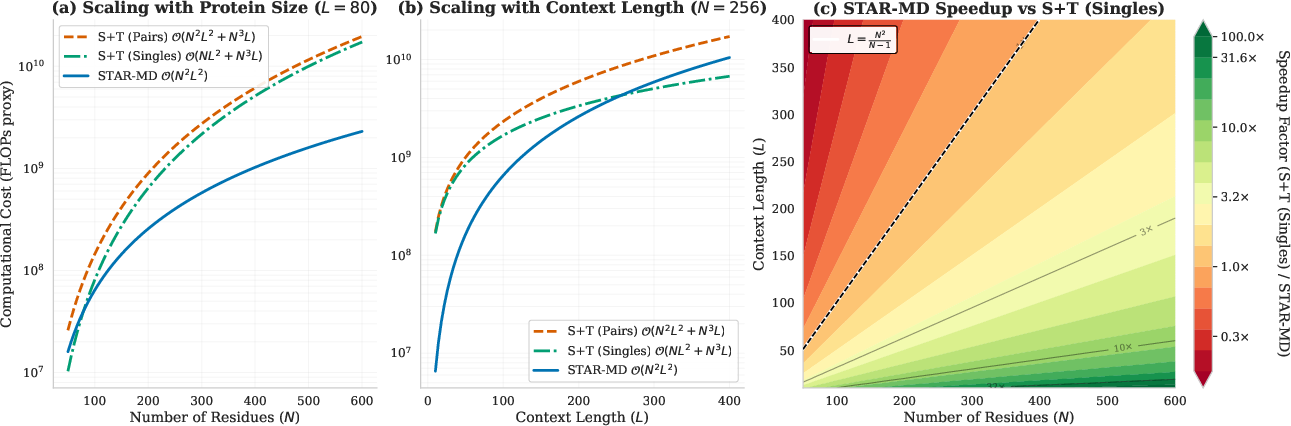
- Efficient for bigger proteins: Because it avoids memory-hungry tricks used by older methods, STAR-MD scales better and can handle longer histories without running out of memory.
Why this matters:
- Many important protein changes happen over longer timescales (microseconds and beyond). Being able to reach those times reliably with an AI model opens the door to studying functionally important motions much faster than standard MD alone.
What could this lead to?
- Faster discovery: Researchers could explore how proteins change shape when they bind to drugs, switch states, or carry signals—much faster and at larger scales.
- Better design: It could help in designing drugs or proteins by quickly predicting how a protein may flex or move in response to changes.
- Smarter simulations: STAR-MD doesn’t replace physics-based MD, but it can complement it—helping guide where to look, filling in gaps, or quickly testing ideas before running very long and expensive simulations.
The bottom line
STAR-MD is like a skilled, physics-aware animator for proteins. It makes long, stable, and realistic movies of protein motion by:
- respecting the geometry of molecules,
- looking at space and time together,
- and learning to avoid error build-up during long animations.
It beats previous methods on standard tests and stays strong even at microsecond scales. This is a big step toward faster, scalable tools for understanding how proteins really move and function.
Knowledge Gaps
Unresolved knowledge gaps, limitations, and open questions
The following list summarizes concrete gaps and open questions that remain after this work and can guide future research:
- Physical consistency of dynamics: The paper does not assess whether generated trajectories satisfy thermodynamic and kinetic principles (e.g., detailed balance, stationary distributions, free-energy surfaces, rate constants). Establishing MSMs/implied timescales and computing transition rates/free-energy profiles from STAR-MD trajectories would test physical fidelity beyond geometric checks.
- Lack of solvent and environment modeling: Trajectories are generated without explicit solvent, membrane, or ionic conditions. Quantify how omission of solvent and environmental variables (temperature, pH, salt, crowding) affects dynamics, and explore conditioning on these variables or hybrid models that couple generative dynamics with environment-aware energy terms.
- Limited system scope (single-chain proteins): The model and evaluations target (apparently) single-chain proteins without ligands or nucleic acids. Extend to multimers, protein–protein/RNA/DNA complexes, and membrane proteins, and evaluate whether joint spatio-temporal attention remains scalable and accurate in these settings.
- Generalization across datasets/force fields: Training and evaluation rely on ATLAS and MD references with a specific force field/protocol. Test robustness across different MD datasets, force fields, and experimental data (cryo-EM ensembles, NMR relaxation), and quantify domain shift effects.
- Long-horizon scalability bounds: Although STAR-MD reaches 1 μs, the O(N2 L2) joint attention may still be prohibitive for very large N or millisecond horizons. Provide empirical scaling curves (runtime, memory, throughput) for protein sizes beyond those tested and horizons >>1 μs, and investigate sparse/structured attention or low-rank approximations.
- Initialization sensitivity: The model conditions on a starting conformation; its robustness to different initial structures (e.g., AlphaFold predictions, homology models, experimental structures) is not tested. Systematically quantify sensitivity to initialization and strategies to mitigate bias toward the starting state.
- Effect of context length and memory modeling: Theoretical motivation (Mori–Zwanzig) implies non-Markovian memory, but empirical characterization of required context length is missing. Vary context length during inference to measure how memory truncation impacts kinetics/coverage and identify minimal effective history windows.
- Optimal contextual noise schedule: The contextual noise perturbation (τ∼U[0,0.1]) improves stability, but the choice of distribution/schedule is ad hoc. Explore principled tuning (e.g., curriculum on τ, state-dependent τ, adaptive τ during inference) and quantify the trade-off between robustness and kinetic fidelity.
- Positional encoding extrapolation limits: 2D RoPE supports some extrapolation, but its stability over extremely long sequences (thousands of frames) and very large residues is not quantified. Benchmark alternative encodings (ALiBi, learned sinusoidal, dynamic relative bias) and measure degradation for L≫training length.
- Impact of removing explicit pairwise features: The “memory inflation” proposition is theoretical; empirical validation is missing. Quantify how omitting pair features affects long-range contact accuracy, allosteric coupling, and residue–residue correlation structure, and test hybrid designs (limited pair features or learned contact priors) to balance cost vs. accuracy.
- Energy-based validation: Structural validity is assessed via MolProbity and clashes/breaks, but no energy/force evaluations (e.g., minimized energy, steric/bond-angle/dihedral energy distributions) are reported. Assess energetic plausibility via force-field scoring or short MD relaxation and measure structural drift.
- Kinetic metrics beyond tICA/VAMP-2: The evaluation lacks MSM construction, implied timescales, Chapman–Kolmogorov tests, and transition-path analyses. Build MSMs from generated trajectories, compare slow-process timescales to MD, and test whether trajectories reproduce metastable state connectivity and lifetimes.
- Rare-event coverage: While recall/JSD are reported, the model’s ability to sample rare but biologically relevant transitions (e.g., folding, large allosteric shifts) is not demonstrated. Design benchmarks focusing on rare events and quantify enrichment, transition-path ensemble similarity, and barrier crossing frequencies.
- Control over “temperature”/variability: The model’s sampling stochasticity is not calibrated to physical temperature or diffusivity. Explore conditioning or sampling controls that tune dynamical variability (e.g., temperature-like parameters) and validate the correspondence to physical diffusion/mobility.
- Side-chain and hydrogen-bond network dynamics: All-atom validity is checked, but dynamic properties of side chains (rotamer kinetics), hydrogen-bond lifetimes, salt bridges, and secondary-structure stability are not analyzed. Include time-resolved residue-level metrics and contact/hydrogen-bond survival analyses.
- Protein size and architecture limits: ConfRover fails on large proteins; STAR-MD claims better scalability, but upper bounds (residue count, attention width) are not fully characterized. Provide comprehensive scaling studies (N from small to very large) and architectural ablations (hidden size, number of layers) to define safe operating regions.
- Runtime and resource reporting: The paper discusses memory but not end-to-end training/inference throughput, wall-clock times, and hardware budgets. Report standardized performance metrics (steps/sec, frames/sec, energy consumption) to enable practical adoption and comparison.
- Conditioning on external perturbations: The framework does not support events like ligand binding/unbinding, mutations, post-translational modifications, or pH jumps. Extend conditioning to such perturbations and evaluate whether STAR-MD can model induced conformational changes and kinetics realistically.
- Uncertainty quantification and calibration: No per-frame or per-transition uncertainty estimates are provided. Develop uncertainty measures (e.g., ensemble sampling, epistemic/aleatoric decompositions) and calibrate them against MD variability.
- Integration with MD (hybrid workflows): It is unclear how STAR-MD could be used to accelerate MD (e.g., as proposal generators, adaptive sampling, seeding MSM states). Design hybrid protocols and quantify speedups vs. fidelity trade-offs.
- Theoretical guarantees and error bounds: Beyond qualitative Mori–Zwanzig arguments, there are no formal guarantees on approximation quality, stability, or error accumulation. Derive error bounds for the learned reverse process and conditions under which joint spatio-temporal attention approximates the memory kernel adequately.
- Dataset selection and evaluation breadth: Long-horizon tests use 32 and 8 proteins; potential selection bias is unaddressed. Expand long-horizon evaluation to broader, systematically sampled protein sets covering diverse folds, sizes, and flexibility regimes.
- Robustness to sequence variations and OOD proteins: The model’s behavior under mutations or for proteins from families absent in training is not tested. Conduct out-of-distribution generalization experiments (mutational scans, novel folds) and measure degradation patterns.
- Reproducibility and ablation completeness: Some architectural choices (e.g., frozen OpenFold features, S×T placement, AdaLN conditioning) are ablated partially; more exhaustive ablations (feature sources, conditioning modalities, attention sparsity) and open-source code/data would strengthen reproducibility and isolate causal contributions.
Practical Applications
Immediate Applications
The following items outline concrete ways the paper’s findings and methods can be deployed now, with sector links, candidate tools/workflows, and feasibility notes.
- Protein ensemble generation for structure-based drug discovery (Sector: healthcare/pharma)
- Use STAR-MD to rapidly generate microsecond-scale conformational ensembles to uncover cryptic/allosteric pockets and conformational states for ensemble docking.
- Workflow: STAR-MD → frame filtering by validity/geometry → pocket detection (e.g., fpocket) → ensemble docking (e.g., AutoDock Vina, Glide) → rescoring.
- Assumptions/dependencies: ligand-free modeling; force-field biases in training MD (ATLAS) propagate; no explicit solvent/ligand conditioning in baseline model; validation against experimental structures still required.
- Triage and prioritization of hit compounds via dynamics-aware ranking (Sector: healthcare/pharma)
- Rank hits by accessibility/occupancy of binding-competent states generated by STAR-MD; prioritize chemistries stabilizing desired conformations.
- Tools: MSM/reweighting over STAR-MD frames; free-energy surrogates; rescoring with physics-based short MD refinement.
- Assumptions: generated populations approximate kinetics qualitatively but not quantitatively; absolute ∆G/kinetics need physics-based follow-up.
- Protein engineering pre-screen for dynamic stability and flexibility (Sector: biotech/biomanufacturing)
- Compare dynamics of wild-type vs. point mutants for loop flexibility, gate opening, and stability before wet-lab assays.
- Workflow: mutate sequence → re-run STAR-MD from (predicted) structure → compare ensemble RMSD, autocorrelation, tICA/VAMP-2 features.
- Assumptions: generalization to mutants depends on training diversity; does not model cofactor/ligand effects unless conditioned or co-modeled.
- Support for structural biology interpretation (cryo-EM/NMR) (Sector: academia/healthcare)
- Generate plausible transition pathways between static structures; reconcile NMR relaxation or cryo-EM class variability with model-predicted intermediates.
- Tools: projection onto experimental PCs/tICA; fitting to low-res maps; Rama/rotamer checks already embedded in evaluation.
- Assumptions: needs careful map/state alignment; no direct conditioning on density maps included (extension needed for map-constrained generation).
- Accelerated MD workflows through hybrid simulation (Sector: academia/pharma)
- Use STAR-MD to seed diverse starting conformations for short, targeted MD or enhanced sampling (e.g., metadynamics), reducing time-to-discovery.
- Workflow: STAR-MD (coarse stride) → select diverse valid frames → short MD refinement → MSM building.
- Assumptions: coarse-grained diffusive steps may miss fine local equilibration; downstream physics-based correction advised.
- Cost-effective, scalable trajectory generation on standard GPUs (Sector: software/cloud/HPC)
- Deploy STAR-MD inference (single-feature KV cache only) for long rollouts on lab GPUs; reduce cloud compute spend vs. large Pairformer backbones or long MD.
- Tools: containerized REST service; scheduling on shared GPU clusters; batching across proteins with block-causal inference.
- Assumptions: training remains GPU-intensive; inference efficiency depends on sequence length and required horizon.
- Data augmentation for representation learning and operator models (Sector: ML/software)
- Generate long, valid trajectories to pretrain encoders (tICA/VAMP-aware) or to train lagged transport/operator models with improved coverage.
- Tools: self-supervised objectives on STAR-MD frames; curriculum with variable strides via continuous-time conditioning.
- Assumptions: synthetic data must be mixed with real MD to avoid model echo-chambers; careful validation against held-out MD.
- Generalizable spatio-temporal modeling components for other domains (Sector: robotics/software/materials)
- Reuse block-causal diffusion, contextual noise perturbation, and joint spatio-temporal attention in SE(3)-like trajectory problems (e.g., robot arm pose forecasting, polymer/RNA dynamics).
- Tools: port 2D-RoPE + causal diffusion to other tokenizations; integrate with task-specific symmetries.
- Assumptions: SE(3) and token definitions differ across domains; retraining required; physics constraints may change.
- Educational visualization of protein dynamics (Sector: education/outreach)
- Create interactive lessons showing conformational landscapes and time-dependent transitions at microsecond scales.
- Tools: web apps linking STAR-MD frames to PCA/tICA plots; validity overlays and stereochemistry dashboards.
- Assumptions: for pedagogy, not for decision-grade predictions; precomputed examples preferred for classroom use.
- Benchmarking and method evaluation extensions (Sector: academia/software)
- Use the paper’s evaluation suite (validity, coverage, tICA/VAMP, autocorrelation) as a standardized benchmark for new trajectory models.
- Tools: publish scripts/datasets; integrate into CI for model regression testing.
- Assumptions: metric choice biases; alignment to MD references needed; requires transparent dataset licensing.
Long-Term Applications
These opportunities require additional research, domain integration, or scaling beyond current capabilities demonstrated in the paper.
- Quantitative kinetics and residence time prediction (k_on/k_off) (Sector: healthcare/pharma)
- Couple STAR-MD ensembles with binding/unbinding event modeling to estimate kinetic parameters driving efficacy and residence time.
- Potential products: SaaS for kinetics-aware lead optimization; kinetic triage dashboards.
- Dependencies: conditioning on ligand presence and concentration; accurate rare-event sampling (≥ millisecond timescale); calibration to experimental kinetics.
- Allosteric modulator and conformational control design (Sector: healthcare/pharma)
- Design small molecules/biologics that shift conformational equilibria to therapeutic states by targeting dynamic allosteric networks.
- Workflow: STAR-MD ensemble → network analysis of coupled motions (joint S×T features) → design cycles with docking/MD validation.
- Dependencies: robust mapping from dynamics to functional outcomes; integration with free-energy methods and experimental feedback.
- De novo enzyme and switchable protein design with dynamic constraints (Sector: biotech/biomanufacturing/energy)
- Engineer catalytic loops and gating motions; design conformational switches responsive to stimuli (pH, ligands, light).
- Products: biocatalysts for green chemistry and biofuels; biosensors with dynamic gating.
- Dependencies: joint sequence–structure–dynamics modeling; co-training with design models (e.g., protein LLMs); experimental iteration.
- Variant effect prediction informed by dynamics (Sector: healthcare/genomics)
- Predict pathogenicity of missense variants by quantifying disruption to dynamic modes and stability landscapes.
- Products: clinical decision support modules enhancing static-structure scores.
- Dependencies: clinical validation cohorts; integration with population genetics and AlphaFold-like static predictors; regulatory acceptance.
- Integrative modeling with experimental constraints (Sector: academia/healthcare)
- Condition generation on cryo-EM maps, NMR restraints, or smFRET data to produce experiment-consistent trajectories.
- Products: map-/restraint-conditional generative pipelines for time-resolved structural biology.
- Dependencies: extend conditioning interfaces; differentiable likelihoods for experimental observables; co-training on multimodal data.
- Physics-informed or hybrid generative–simulation platforms (Sector: software/HPC)
- Embed differentiable physics constraints or couple to short MD steps for stable long-horizon generation at millisecond scales.
- Products: hybrid engines offering user-selectable speed–fidelity trade-offs.
- Dependencies: scalable differentiable modules; stability analysis; standardized APIs across MD packages (OpenMM, GROMACS).
- Molecular nanotechnology and dynamic biomaterials design (Sector: materials/energy/robotics)
- Design dynamic protein assemblies and molecular machines with prescribed motion cycles for targeted transport, catalysis, or energy transduction.
- Dependencies: reliable prediction beyond microseconds; co-modeling of multi-protein complexes and solvent/ion effects; manufacturing constraints.
- Policy and sustainability frameworks for compute-efficient biosimulation (Sector: policy/HPC/ESG)
- Inform funding and infrastructure allocation by quantifying energy/CO₂ savings from generative acceleration vs. brute-force MD.
- Products: best-practice guidelines for low-carbon discovery pipelines; benchmarks for reproducibility and validation.
- Dependencies: standardized reporting; cross-institution validation; lifecycle assessments of training vs. inference costs.
- Enterprise platforms for dynamics-first discovery (Sector: software/SaaS/pharma)
- Integrated platforms offering secure, scalable STAR-MD inference, ensemble analytics, and workflow orchestration with ELNs/LIMS.
- Features: project templates (cryptic-pocket discovery, allostery scans), governance, audit trails, and API integrations.
- Dependencies: robust MLOps; IP/security requirements; SLAs for regulated environments; continuous model validation.
- Cross-domain transfer of joint spatio-temporal diffusion (Sector: robotics/autonomy)
- Apply causal, joint S×T diffusion with contextual noise to long-horizon motion planning and multi-agent forecasting in SE(3).
- Products: planners predicting long-horizon trajectories with reduced error accumulation.
- Dependencies: domain-specific symmetries and constraints; safety and interpretability standards; large-scale task-specific datasets.
Key cross-cutting assumptions and dependencies
- Training data bias: learned dynamics reflect the MD force fields, water models, and sampling biases in ATLAS and extended simulations; extrapolation to new classes (membrane proteins, complexes) may require domain-specific training.
- Environment and ligands: current setup focuses on apo proteins; many impactful applications require explicit ligand, cofactor, membrane, or solvent conditioning.
- Timescale limits: demonstrated stability to microseconds; rare-event and millisecond phenomena remain research challenges.
- Validation: structural validity ≠ biochemical function; experimental corroboration and physics-based refinement remain essential for high-stakes use.
- Compute: while inference is efficient, training is resource-intensive; enterprise deployment needs MLOps and governance for versioning and reproducibility.
Glossary
- AdaLN (Adaptive LayerNorm): A conditioning mechanism that modulates layer activations based on auxiliary inputs such as time; enables the network to adapt to varying timescales. "through adaptive layernorm (AdaLN, \cite{peebles2023scalable}),"
- Allosteric regulation: Modulation of a protein’s activity by binding at a site distinct from the active site, often inducing conformational changes. "allosteric regulations"
- ATLAS dataset: A benchmark collection of protein molecular dynamics trajectories used for training and evaluation. "ATLAS dataset~\citep{vander2024atlas} contains \SI{100}{\nano\second} MD trajectories."
- Autocorrelation: A measure of how much a signal or trajectory is correlated with itself at different time lags, used to assess temporal memory. "autocorrelation to assess temporal memory,"
- Autoregression: A generation paradigm where each new frame is produced conditioned on previously generated frames. "LLM-style autoregression"
- Cα superposition: Structural alignment method using alpha-carbon atoms to superimpose protein conformations. "via C superposition prior to analysis (\cref{ap:eval})."
- Cα–Cα clashes: Non-physical overlaps where alpha-carbons get too close, indicating steric clashes. "C-C clashes (non-bonded atoms too close)"
- Cβ distance: The distance between beta-carbon atoms of residues, often used as a pairwise geometric feature. "pairwise distance within each frame"
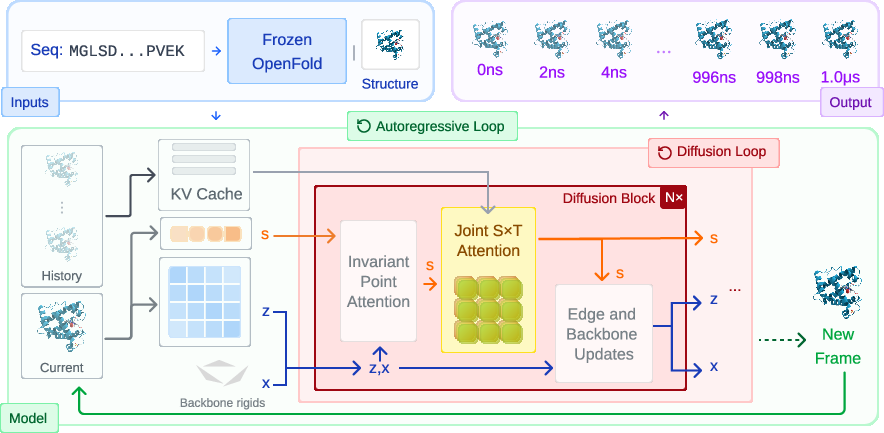
- Causal diffusion transformer: A transformer-based diffusion architecture with causal attention for autoregressive conditioning over time. "a causal diffusion transformer with joint spatio-temporal attention"
- Chain breaks: Artifacts where consecutive residues are too far apart, indicating discontinuities in the protein backbone. "chain breaks (consecutive C atoms too far apart)"
- Denoising score matching: A training objective that learns the gradient of the data log-density to denoise corrupted inputs in diffusion models. "is trained via denoising score matching"
- Diffusion Forcing: A technique that perturbs historical context with small diffusion noise to improve robustness in long rollouts. "inspired by Diffusion Forcing~\citep{chen2024diffusion, song2025history}."
- EdgeTransition layer: An architectural module that updates pairwise features from single-residue features. "using the EdgeTransition layer from \citet{jumper2021_af2}."
- Equivariant (SE(3)-equivariant): A property where model outputs transform consistently under rotations and translations in 3D space. "an -equivariant diffusion model"
- Generalized Langevin Equation: A formalism describing coarse-grained dynamics with memory and stochastic terms arising from eliminated degrees of freedom. "yields a Generalized Langevin Equation with three terms"
- IGSO3 (Isotropic Gaussian on SO(3)): A distribution over 3D rotations used to diffuse rotational components. "IGSO is the isotropic Gaussian on ,"
- Invariant Point Attention (IPA): An attention mechanism that respects rotational and translational invariances for geometric inputs. "Invariant Point Attention layers \citep{jumper2021_af2} update single features"
- Jensen-Shannon Divergence (JSD): A symmetric measure of divergence between probability distributions used to assess coverage. "compute the Jensen-Shannon Divergence (JSD)"
- Joint Spatio-Temporal Attention (S×T): An attention mechanism operating on residue–frame tokens to capture non-separable space-time dependencies. "Joint Spatio-Temporal Attention."
- KV cache (Key–Value cache): Stored attention keys and values enabling efficient autoregressive inference with long contexts. "via KV-caching."
- LogUniform distribution: A distribution where the logarithm of the variable is uniformly distributed, used to sample physical stride across scales. "\,ns"
- Markovian: A property of processes where the future state depends only on the current state, not on past history. "approximate evolution as a Markovian process,"
- MolProbity: A suite for detailed all-atom structural validation, assessing stereochemical quality. "we use the MolProbity suite~\citep{chen2010molprobity}"
- Mori–Zwanzig formalism: A projection-operator framework that derives effective dynamics with memory terms for coarse-grained variables. "we use the Mori-Zwanzig formalism~\citep{mori1965transport, zwanzig1961memory}"
- Non-Markovian: Dynamics where future states depend on past history, often due to coarse-graining or partial observation. "fail to capture the non-Markovian properties"
- Pairformer: An architectural backbone that processes single and pairwise features with specialized operations like triangular attention. "Pairformer~\citep{jumper2021_af2} backbone"
- Principal components (PCs): Orthogonal directions of maximal variance used to project and visualize conformational landscapes. "onto the first two principal components (PCs) of the reference MD simulation"
- Ramachandran: The distribution of backbone dihedral angles (ϕ, ψ); used to identify stereochemical outliers. "backbone Ramachandran and side-chain rotamer outliers"
- Riemannian manifold: A curved space with a smoothly varying metric; SE(3) is treated as such for diffusion over poses. "on the Riemannian manifold ,"
- Rotary Position Embedding (RoPE): A positional encoding method enabling extrapolation, extended here to 2D for residue and frame indices. "2D Rotary Position Embedding (2D-RoPE)~\citep{heo2024rotary}"
- Rotamer: A discrete side-chain conformation defined by preferred dihedral angles; outliers indicate invalid stereochemistry. "side-chain rotamer outliers"
- SE(3): The group of 3D rigid motions (rotations and translations), used to represent backbone rigid coordinates. ""
- SO(3): The group of 3D rotations; rotational components of structures reside in this manifold. ""
- tICA (time-lagged independent component analysis): A method to extract slow collective variables by maximizing autocorrelation at a lag. "tICA lag-time correlation"
- Torsional angles: Dihedral angles around bonds, especially in side chains, defining detailed conformations. "with torsional angles for side-chain atoms."
- Transport operators: Learned mappings that evolve structures to future times, typically at fixed lags. "aim to learn transport operators that predict conformations at lagged times."
- Triangular attention: A specialized attention operation over pairwise features used in AlphaFold2-like architectures. "AlphaFold2-style triangular attention"
- VAMP-2 score: A metric from the variational approach for Markov processes quantifying the quality of learned slow dynamics. "and VAMP-2 score to evaluate how well slow dynamical modes of the system are captured."
Collections
Sign up for free to add this paper to one or more collections.