Mechanisms of AI Protein Folding in ESMFold
Abstract: How do protein structure prediction models fold proteins? We investigate this question by tracing how ESMFold folds a beta hairpin, a prevalent structural motif. Through counterfactual interventions on model latents, we identify two computational stages in the folding trunk. In the first stage, early blocks initialize pairwise biochemical signals: residue identities and associated biochemical features such as charge flow from sequence representations into pairwise representations. In the second stage, late blocks develop pairwise spatial features: distance and contact information accumulate in the pairwise representation. We demonstrate that the mechanisms underlying structural decisions of ESMFold can be localized, traced through interpretable representations, and manipulated with strong causal effects.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
A simple explanation of “Mechanisms of AI Protein Folding in ESMFold”
What is this paper about?
This paper asks a big question: how does an AI model turn a string of amino acids (the building blocks of proteins) into a 3D shape? The authors look inside a popular model called ESMFold to see how it decides on a shape, focusing on a small but important protein shape called a beta hairpin (two short strands running side by side with a tiny loop that turns the chain around).
What questions are the researchers trying to answer?
To make the model’s “thinking” easier to understand, the authors focus on questions like:
- When, during the model’s step-by-step process, does it decide key parts of the shape?
- What information does it use early on versus later on?
- Can we nudge or change the model’s internal “notes” and make it change the predicted shape on purpose?
How did they study it? (Methods in everyday language)
Think of ESMFold as a machine with two kinds of notebooks it uses while working:
- A per-letter notebook (sequence notes): one note for each amino acid in the chain.
- A big grid notebook (pair notes): a table that stores information about every possible pair of amino acids (like who might be near whom, or who might interact).
The model goes through many steps (called blocks). Early steps and late steps might do different things.
The authors used three main tricks:
- Copy-and-paste tests (activation patching): They took internal notes from a “donor” protein that has a beta hairpin and pasted those notes into a “target” protein that normally forms helices. If the target suddenly forms a hairpin, that shows those notes caused the change. They tried pasting either the per-letter notes or the pair notes and did this at different steps to see when each type matters.
- Steering a dial (feature steering): They found simple “directions” in the model’s notes that mean real chemistry, like electrical charge (positive vs. negative). Pushing the notes along these directions is like turning a dial toward “more positive” or “more negative,” then watching how the predicted shape changes.
- Distance reading and nudging (probing and steering): They trained a simple tool to read distances between amino acids directly from the pair notes (like reading a map). Then they gently nudged those notes to make certain pairs move closer or farther, to see if the model would create the expected bonds.
Along the way, they explained technical ideas with plain terms:
- Beta hairpin: two short “ribbons” (beta strands) side by side, connected by a short loop, stabilized by hydrogen bonds (weak attractions between backbone atoms).
- Pair notes (pairwise representation): a big table that stores the relationship between any two positions in the protein.
- Hydrogen bonds: the “Velcro” that helps hold strands together in sheets.
What did they find, and why does it matter?
The authors discovered that ESMFold’s folding process happens in two stages with different jobs:
- Stage 1 (early steps): The model moves chemistry into the pair notes.
- Here, the model pulls information like residue identity and charge (positive/negative) from the per-letter notes and writes it into the pair notes. This sets up which pairs are likely to interact, but doesn’t yet lock in exact 3D distances.
- Evidence: Copying per-letter notes from a hairpin into a target protein only works early on—it can make the target region form a hairpin. Blocking the route from per-letter notes to pair notes during early steps stops this effect.
- Stage 2 (later steps): The model builds geometry in the pair notes.
- The pair notes now grow strong distance and contact information—basically a map that says which residues should be close.
- Evidence: Copying pair notes from a hairpin into a target protein works best in later steps, making the target form a hairpin-like shape. Simple tests can read distances directly from these pair notes with high accuracy late in the process.
They also showed these ideas are causal, not just correlations:
- Charge steering: Making one side of a potential hairpin more positive and the other more negative pulls them together and increases the formation of the hydrogen bonds that define beta sheets. Making both sides the same charge pushes them apart—exactly what you’d expect from basic chemistry.
- Distance steering: Nudging the pair notes to target the typical cross-strand spacing for beta sheets makes the strands move closer and form more hydrogen bonds.
- Attention guidance: Late in the process, the pair notes send “biases” back to the per-letter notes to tell the model which residues should “talk” to each other more (for example, pairs that should form contacts).
- Global scaling: If you scale the pair notes up or down before the final step, the predicted protein expands or shrinks accordingly. This shows the pair notes act like a geometric blueprint the final module uses to draw the 3D shape. Scaling the per-letter notes doesn’t do this.
Why it matters:
- These findings explain how the model actually builds a 3D shape from a sequence: early chemistry → late geometry.
- They show we can steer models with simple, understandable directions (like charge or distance), which could help fix errors or design proteins on purpose.
What’s the big picture impact?
This work opens the “black box” of AI protein folding and shows:
- We can locate when important shape decisions happen.
- We can identify what the model stores (chemistry first, then geometry).
- We can change the model’s mind in targeted, meaningful ways.
In practice, that could lead to:
- Better debugging when a model gets a structure wrong.
- Safer and more controllable tools for protein design.
- New scientific insights extracted from what these models have learned.
The study focused on one model (ESMFold) and one common shape (beta hairpins), but the methods can be reused to understand other models and other protein features.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
The following list summarizes what remains missing, uncertain, or unexplored in the paper, framed as concrete directions for future research:
- Generalization beyond a single motif: The analysis focuses on beta hairpins and helix-turn-helix regions; it remains unclear whether the two-stage computation (early biochemical propagation, late geometry) holds for other secondary motifs (e.g., parallel β-sheets, β-arches, α/β sandwiches), larger tertiary folds, or full proteins with complex topologies.
- Model generality across architectures: Results are shown only for ESMFold. It is unknown whether the same stage structure, pathways (seq2pair/pair2seq), and representational roles of z replicate in AlphaFold2/OpenFold/RoseTTAFold/Boltz-2 or in multimeric settings.
- Impact of recycling: All causal analyses disable recycling and use a single trunk pass; whether the identified mechanisms persist, shift in timing, or are overwritten across recycles is not evaluated.
- Dependence on short proteins: Experiments are performed on relatively short proteins; scalability to longer chains, domain architectures, and multi-domain proteins is untested.
- Incomplete mechanistic decomposition of the trunk: Triangular multiplicative updates and triangular attention are treated as a black box. Which components (layers/heads/channels) within these updates build distance/contact features, and how, is not localized.
- Structure module internals remain opaque: Aside from scaling tests, the IPA-based structure module is treated as a black box. How specific features of z (e.g., channels, heads, or directions) are read into geometry (rotations, translations, frames) is not mapped.
- Orientation and registry information in z: The work shows linear accessibility of distances from z, but does not probe whether strand orientation (antiparallel vs parallel), registry, chirality, and torsion-level constraints are represented and causally controllable.
- Side-chain geometry and specific interactions: Analyses use Cα distances and backbone H-bonding as proxies; how z encodes side-chain placement, specific H-bond geometries, salt bridges, cation–π, or aromatic stacking remains unexplored.
- Beyond charge: Only charge is investigated as a biochemical feature. Whether hydrophobicity, polarity, β-strand propensity, turn propensity (e.g., proline/glycine effects), size, or aromaticity are linearly encoded, transmitted via seq2pair, and causally influence folding is not tested.
- Linearity assumptions: Probing and steering rely on linear directions in s and z. The extent to which nonlinear structure (e.g., multiple subspaces, feature entanglement, or head-specific nonlinearities) is necessary for folding decisions is not assessed.
- Stability and uniqueness of directions: The consistency of “charge” and “distance” directions across proteins, blocks, seeds, and training checkpoints is untested; whether multiple equivalent or competing directions exist is unknown.
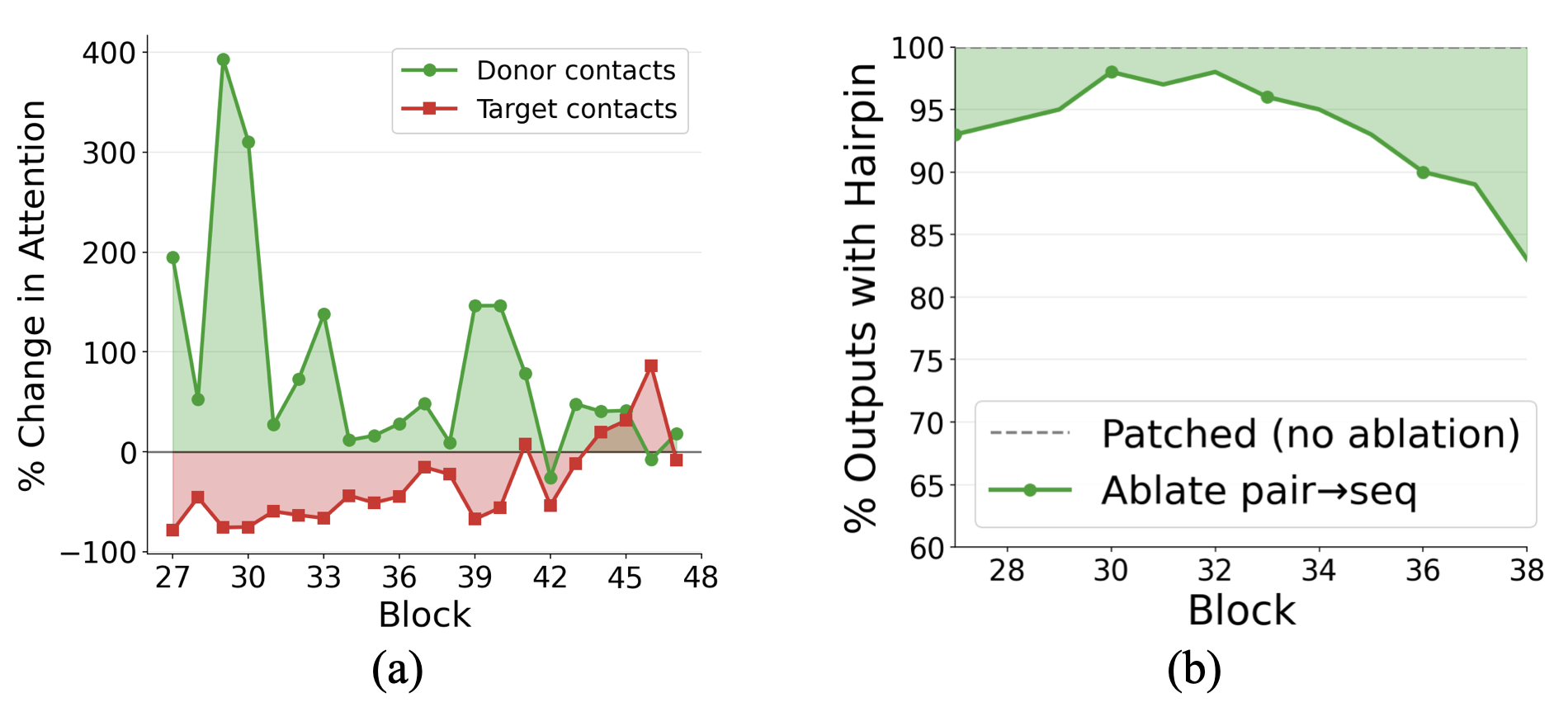
- Pair2seq causal necessity: Ablating pair2seq after z patching reduces hairpin formation only modestly (10–20%), leaving the main causal path(s) from z to structure (beyond IPA) unidentified and unquantified.
- Head specialization and per-head roles: Pair2seq biases suggest head specialization, but per-head functions, contact selectivity, and division of labor within attention are not mapped.
- Patching scope and boundary effects: Patches are applied to localized regions aligned by loops, but how patched regions interact with the unpatched context (e.g., conflicts, boundary artifacts) and how alignment choices affect outcomes are not systematically studied.
- Success/failure determinants: Approximately 40% success under full trunk patching and lower rates for single-block patches leave many failures unexplained; feature-level or sequence-level predictors of patching success are not identified.
- Global compaction confound: Compaction is partially controlled by filtering on radius of gyration, but a comprehensive analysis distinguishing true contact formation from nonspecific collapse across interventions is lacking.
- Confidence and quality metrics: Effects of interventions on model confidence (e.g., pLDDT, PAE) and stereochemical plausibility are not reported, limiting assessment of structure quality under perturbations.
- Temporal precision of stages: The early-window “write-in” period is identified empirically (~blocks 0–10), but the exact block-by-block transition of responsibilities (seq2pair vs triangular updates vs pair2seq) is not mechanistically derived or pinned to specific sublayers.
- Distance steering specificity: Distance steering targets Cα distances (e.g., 5.5 Å) and induces H-bonds, but whether steering can control more detailed geometric objectives (e.g., backbone dihedrals, sheet twist, hydrogen bond angles) is unknown.
- Interaction with long-range/global contacts: The focus is on local hairpin contacts. Whether late-stage z representations orchestrate long-range tertiary packing and how pair2seq biases or triangular updates scale to global topology is not evaluated.
- Robustness to OOD sequences: Generalization of learned directions/probes to out-of-distribution sequences (e.g., low-complexity regions, repeats, designed sequences) is not tested.
- Multimeric interfaces and cofactors: The study addresses monomers without ligands, cofactors, disulfides, or multimeric interfaces; whether similar mechanisms regulate interface contacts or ligand-induced conformations is unexamined.
- Training data and pretraining dependence: How the observed mechanisms depend on ESM-2 pretraining, PDB biases, or training objectives (e.g., distogram heads) is not assessed (e.g., ablations with alternative pretraining or training regimes).
- Registry of causal channels: While z acts as a “distance map,” the mapping between specific z dimensions/channels and geometric features remains unknown, limiting targeted interpretability and design of interventions.
- Design applications: Although steering/patching causally modulates structures, the paper does not translate these findings into sequence design or targeted fold-engineering protocols; a pipeline to convert representational steering into sequence-level edits is absent.
These gaps suggest concrete experiments: replicate analyses in multiple models and motifs; dissect triangular updates and IPA at the head/channel level; extend biochemical steering beyond charge; probe orientation/registry and dihedrals; study recycling dynamics; quantify confidence/sanity checks; and develop principled mappings from latent interventions to actionable design edits.
Practical Applications
Immediate Applications
The following applications can be deployed now, leveraging the paper’s causal analyses, interventions, and diagnostics for ESMFold-like models.
- Mechanistic debugging and QC of structure predictions (Sector: software/biotech R&D)
- Tool/product/workflow: “ESMFold Interpretability Toolkit” that exposes hooks for block-wise activation patching, seq2pair/pair2seq ablations, and z-scaling sensitivity checks to localize failure modes.
- Use cases:
- Pinpoint whether misfolds arise in early biochemical propagation (seq2pair) or late geometric consolidation (z/triangular updates).
- Flag suspect predictions when output geometry is overly sensitive to pairwise scaling or when pair2seq bias fails to separate contacts (low ROC-AUC).
- Assumptions/dependencies:
- Access to model internals (activations and trunk blocks), typically requiring self-hosted ESMFold/OpenFold or instrumented builds.
- Generalization to very long proteins and multi-recycle inferences may require re-tuning windows.
- Motif induction for design prototyping (Sector: biotech/pharma, academic protein engineering)
- Tool/product/workflow: “Motif Induction Assistant” that applies early-block charge steering to promote cross-strand complementarity and late-block distance steering to bring residues into contact.
- Use cases:
- Rapidly bias a helix–turn–helix design toward a beta-hairpin-like arrangement for feasibility checks before full MD or experimental validation.
- Enforce or test presence of specific contacts in de novo peptides.
- Assumptions/dependencies:
- Steering can produce non-physical configurations; downstream energy-based screening (e.g., Rosetta, MD) is recommended.
- Strength/placement of steering windows must be calibrated to avoid global compaction.
- Variant-effect triage via stage-aware diagnostics (Sector: healthcare, precision medicine)
- Tool/product/workflow: A variant analysis plugin that probes whether mutations act through early biochemical features (e.g., charge) or late geometric contacts.
- Use cases:
- Prioritize mutations that alter charge/projected contact bias in critical motifs, informing experimental testing.
- Assumptions/dependencies:
- Requires validation against clinically annotated datasets; best used as a heuristic prioritization signal, not a standalone clinical decision tool.
- Template and constraint injection tuned to trunk stages (Sector: software for structural biology)
- Tool/product/workflow: Enhanced template-conditioning that injects constraints directly into z during late blocks, or uses linear distance probes to set soft targets.
- Use cases:
- Improve template-based modeling by synchronizing when and where constraints are applied for maximal effect.
- Assumptions/dependencies:
- Distance probes trained on one model/version may need retraining for others.
- Structure reliability scoring beyond pLDDT (Sector: biotech R&D)
- Tool/product/workflow: QC dashboard combining (a) contact-bias ROC from pair2seq, (b) linear distance probe R² over blocks, and (c) z-scaling sensitivity.
- Use cases:
- Filter low-confidence folds by internal-mechanism consistency, not only final output scores.
- Assumptions/dependencies:
- Calibrations needed per protein class (e.g., membrane vs. soluble).
- Teaching and visualization (Sector: education, citizen science)
- Tool/product/workflow: Interactive demos showing early-stage charge propagation and late-stage distance/contact emergence; overlays of pair2seq biases and z-distance readouts.
- Use cases:
- Educate students and citizen scientists (e.g., Foldit-like platforms) about how AI folding models make structural decisions.
- Assumptions/dependencies:
- Simplifications are advisable for non-expert audiences; restrict steering ranges to safe regions.
- Architecture-informed training diagnostics (Sector: ML engineering for bio)
- Tool/product/workflow: Training-time monitors for seq2pair contribution profiles, block-wise distance probe R², and contact-bias development to detect regressions.
- Use cases:
- Catch early/late-stage undertraining issues during model development.
- Assumptions/dependencies:
- Requires access to training loop and overhead for extra probes/metrics.
- Open-source reproducibility and benchmarking (Sector: academia)
- Tool/product/workflow: Public reproducible benchmarks that report how well new models exhibit the two-stage regime and respond to charge/distance steering.
- Use cases:
- Standardize interpretability-oriented comparisons between folding models.
- Assumptions/dependencies:
- Community agreement on datasets and metrics; consistent hardware/software environments.
Long-Term Applications
These opportunities require further research, model changes, scale-up, and validation across motifs, protein classes, and tasks.
- Controllable folding models with user-specified constraints (Sector: biotech/pharma)
- Tool/product/workflow: “Constraintable ESMFold” that exposes APIs to set block-local steering for motif presence, distance targets, and contact maps; integrated solvers to find minimal steering achieving target geometry.
- Prospective products:
- Design IDEs where users dial in motifs (e.g., hairpins, beta-sheets) and contact patterns, then export candidates for energy-based refinement.
- Assumptions/dependencies:
- Must demonstrate physical plausibility and robust constraint satisfaction across long proteins and complexes; safety/capability governance required.
- Closed-loop generative design with interpretable feedback (Sector: protein design, materials)
- Tool/product/workflow: Integration with RFdiffusion/other generative models to (i) propose sequences, (ii) apply stage-aware steering to meet structural criteria, (iii) score via interpretability-QC, (iv) iterate.
- Use cases:
- De novo fold creation, enzyme active site pre-organization, materials with target beta-sheet content (e.g., silk-like fibers).
- Assumptions/dependencies:
- Requires cross-model alignment and calibrations; extensive experimental validation.
- Mechanism-informed mutational rescue and rational engineering (Sector: therapeutics, antibodies, enzymes)
- Tool/product/workflow: Automated design suggestions that propose compensatory mutations guided by early (charge) vs. late (contact) deficits.
- Use cases:
- Stabilize unstable motifs in antibodies (e.g., beta-rich frameworks), reintroduce key contacts in engineered enzymes.
- Assumptions/dependencies:
- Complex epistasis and allostery may confound localized steering; empirical screening still necessary.
- Interpretable multi-chain docking and complex assembly control (Sector: structural biology, biologics)
- Tool/product/workflow: Extensions of z-level distance/contact steering to multi-chain interfaces; pair2seq-guided attention shaping across chains.
- Use cases:
- Rationally induce or disrupt interfaces; enforce geometry in multispecifics and scaffolds.
- Assumptions/dependencies:
- Requires extension and validation on complex/docking models; potential interactions with training biases.
- Regulatory standards and audits for explainable bio-AI (Sector: policy/governance)
- Tool/product/workflow: Audit frameworks requiring evidence that folding predictions pass mechanism-based checks (e.g., stable contact bias, consistent distance encoding) before use in high-stakes pipelines.
- Use cases:
- Improve accountability, detect out-of-distribution behaviors, and reduce inadvertent misuse.
- Assumptions/dependencies:
- Community consensus and standard-setting; balancing transparency with IP and security concerns.
- Safety controls and capability gating (Sector: policy, platform security)
- Tool/product/workflow: Platform-level throttles on controllability (e.g., limits on steering strength/windows) and mandatory interpretability logs for sensitive projects.
- Use cases:
- Mitigate dual-use risk while allowing beneficial R&D.
- Assumptions/dependencies:
- Governance processes and secure compute environments; user education.
- Discovery of new biochemical “rules” from model internals (Sector: academia)
- Tool/product/workflow: Systematic mining of linear directions for other biochemical attributes (hydrophobicity, aromaticity, backbone preferences) and their causal roles in folding.
- Use cases:
- Derive, test, and publish interpretable principles that generalize beyond training data, informing textbooks and curricula.
- Assumptions/dependencies:
- Extensive cross-motif, cross-model validations and experiments; risk of over-interpreting model artifacts.
- Cross-domain transfer of stage-aware interpretability patterns (Sector: ML software/engineering)
- Tool/product/workflow: Apply “early semantics → late geometry” design principles to other pair-structured models (e.g., RNA folding, chromatin contact prediction).
- Use cases:
- Guide architecture and interpretability in adjacent structural biology tasks.
- Assumptions/dependencies:
- Biological differences may invalidate direct transfer; bespoke analyses needed.
- Industrial-scale, model-agnostic interpretability services (Sector: bioinformatics software)
- Tool/product/workflow: Cloud services that run standardized interpretability batteries (patching, probes, steering, sensitivity) on user-provided sequences across multiple folding models, returning actionable reports.
- Use cases:
- Enable SMEs to adopt interpretability without deep in-house expertise.
- Assumptions/dependencies:
- Licensing and API access to underlying models; secure data handling and compliance.
- Enhanced education and outreach with causal simulations (Sector: education, public engagement)
- Tool/product/workflow: Classroom labs where students apply charge/distance steering and see structural consequences, linking chemistry to 3D outcomes.
- Use cases:
- Foster literacy in AI-driven molecular science and responsible use.
- Assumptions/dependencies:
- Simplified, safe demos; teacher training and curricular integration.
Cross-cutting dependencies and caveats
- Generalization beyond beta-hairpins and ESMFold needs verification for other motifs, long proteins, intrinsically disordered regions, membrane proteins, complexes, and models (e.g., AlphaFold2/OpenFold variants).
- Experiments minimized recycling; behavior under full recycling may differ.
- Steering may yield non-physical minima; always pair with physics-based validation and experimental assays.
- Access to internal activations is required; hosted APIs may not support this without vendor cooperation.
- Biosecurity: added controllability/insight could accelerate both beneficial and harmful applications—governance and access controls are critical.
Glossary
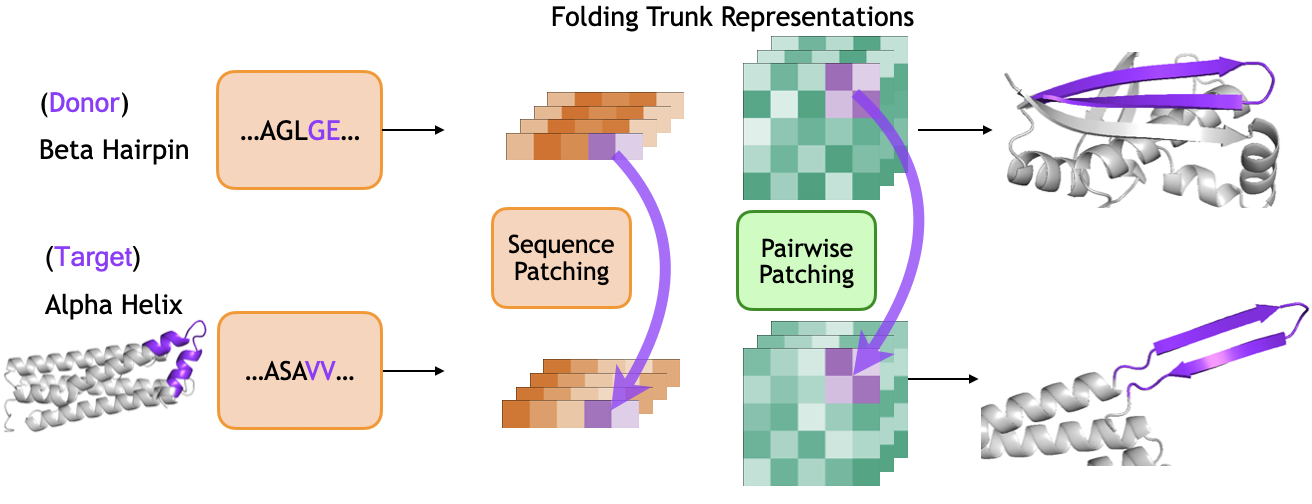
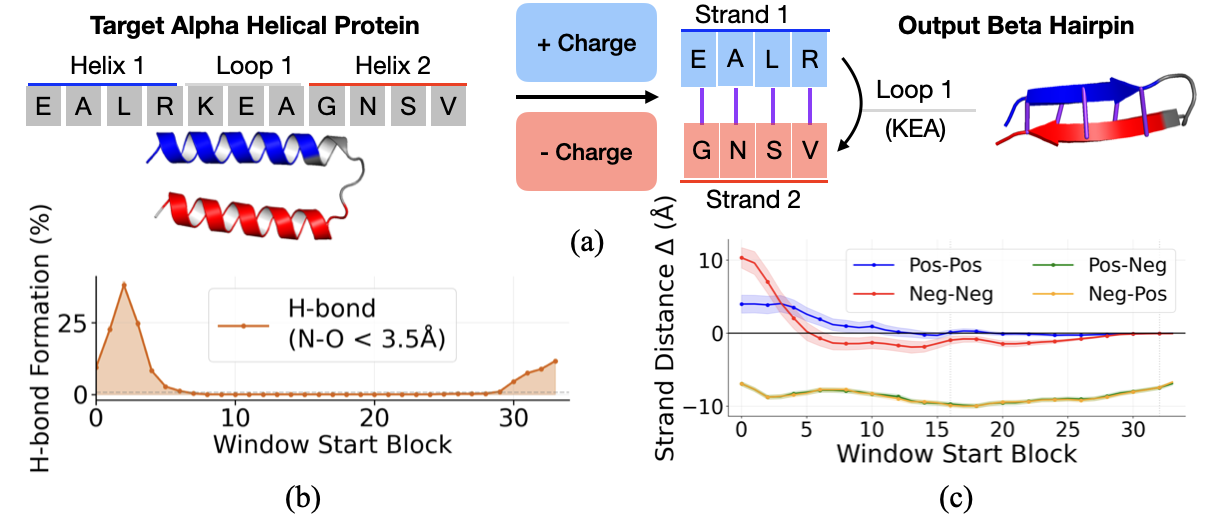
- Activation patching: A causal analysis technique that replaces internal activations from a donor run into a target run to test which representations drive behavior. "We use activation patching \cite{vig2020causal,meng2022locating} to transplant sequence and pairwise representations from the forward pass of a donor protein containing a beta hairpin into the forward pass of a target protein containing an alpha-helical region."
- Alpha-helical: Refers to protein regions forming alpha helices, a common secondary-structure element. "We curated a dataset of 95 target proteins consisting of alpha-helical structures from diverse protein families."
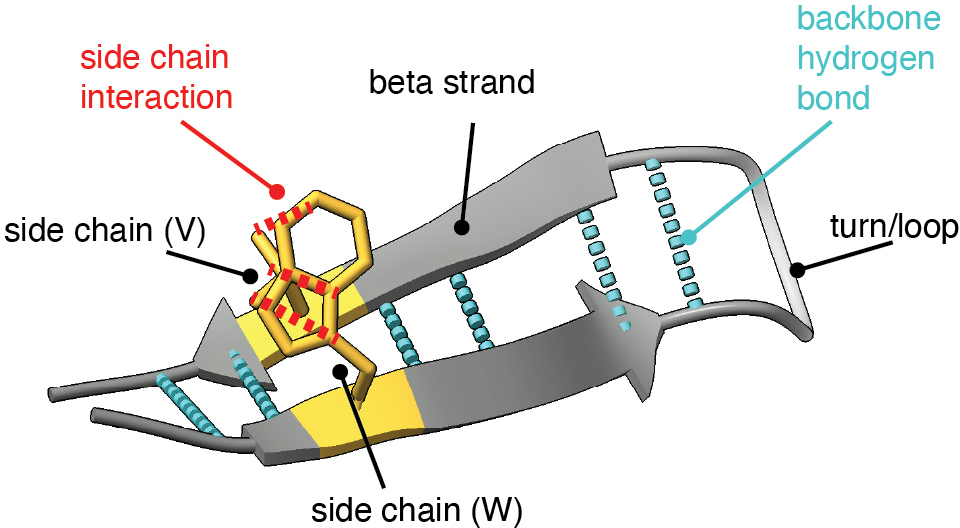
- Antiparallel: Orientation of two strands running in opposite directions along the backbone, typical in beta sheets. "A beta-hairpin (Fig.~\ref{fig:hairpin-secondary}) is a common secondary-structure motif formed by two beta strands that run in opposite directions along the protein backbone (antiparallel orientation)."
- Backbone: The common structural chain of a residue (N–Cα–C=O) that forms the protein’s main chain and participates in hydrogen bonding. "The backbone contains the amino nitrogen (N), central carbon (C), and carbonyl group (C=O); C--C distances are commonly used to measure inter-residue distances, while backbone N--H and C=O groups form the hydrogen bonds that stabilize secondary structures."
- Beta hairpin: A secondary-structure motif of two short antiparallel beta strands connected by a loop. "We investigate this question by tracing how ESMFold folds a beta hairpin, a prevalent structural motif."
- Beta strand: A secondary-structure element that aligns side-by-side with others to form beta sheets. "A beta-hairpin (Fig.~\ref{fig:hairpin-secondary}) is a common secondary-structure motif formed by two beta strands that run in opposite directions along the protein backbone (antiparallel orientation)."
- Boltz-2: A protein structure prediction model architecture related to AlphaFold/OpenFold. "Like AlphaFold2~\citep{jumper2021highly}, OpenFold~\citep{Ahdritz2024-yd}, and Boltz-2~\cite{Passaro2025.06.14.659707}, ESMFold~\citep{esmfold} is built around a folding trunk"
- Cα (C-alpha): The alpha-carbon atom in amino acids; Cα–Cα distances are a standard measure of residue spacing. "C--C distances are commonly used to measure inter-residue distances"
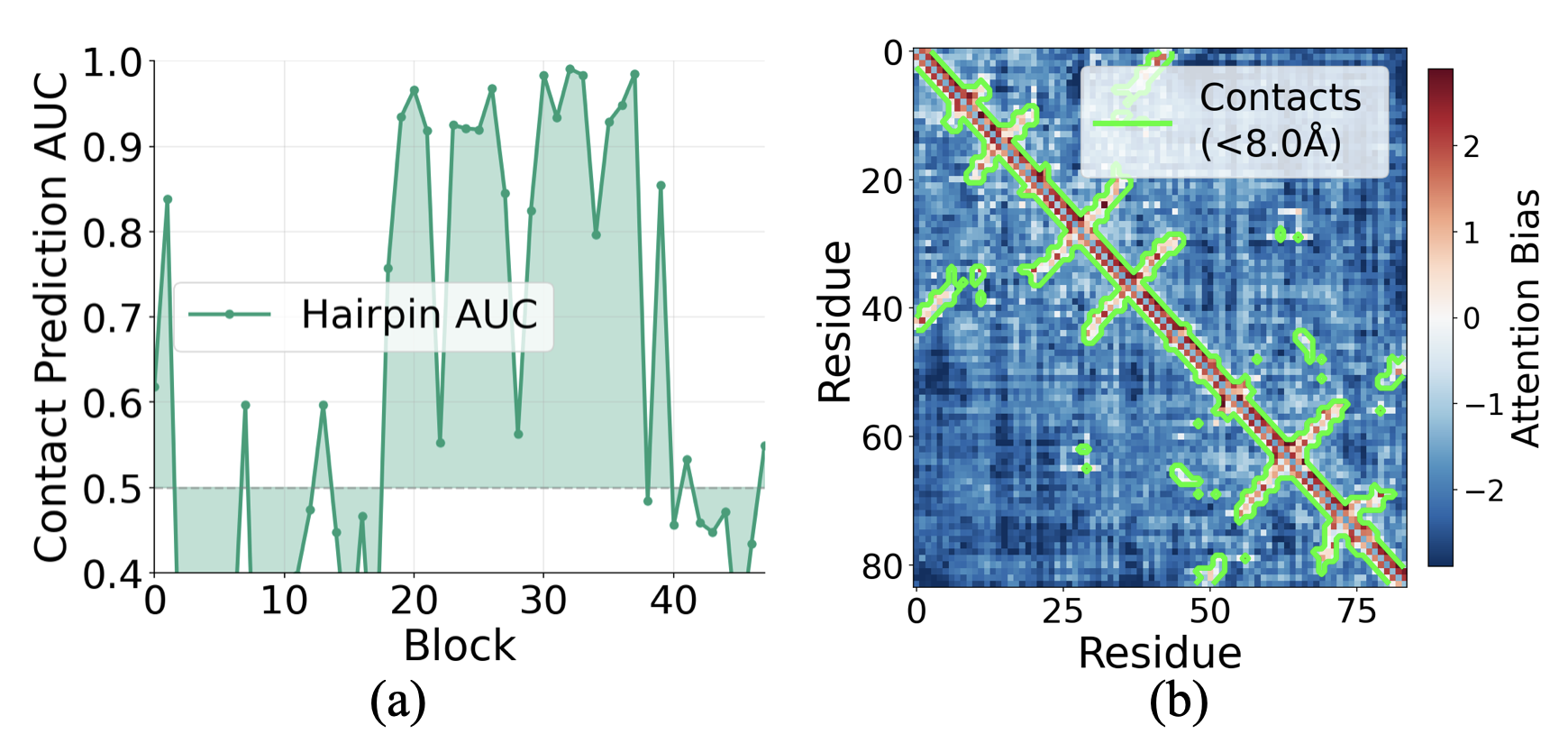
- Contact map: A representation indicating which residue pairs are in spatial proximity (contact). "Visualizing bias values alongside contact maps reveals that residue pairs in contact receive substantially more positive bias than non-contacts (Fig.~\ref{fig:contact_maps}b)."
- Cryo-EM: Cryogenic electron microscopy, an experimental technique for determining 3D structures of biomolecules. "By predicting three-dimensional structures directly from amino acid sequences, neural networks such as AlphaFold and ESMFold often eliminate the need for labor-intensive crystallography~\cite{glusker1996crystal} and cryo-EM~\citep{resolution_revolution} experiments"
- Difference-in-means approach: A method to find a direction in representation space by subtracting mean vectors of two classes. "To test whether charge is encoded, we used a difference-in-means approach to identify a ``charge direction'' in the sequence representation space."
- Distogram head: A model head that predicts binned pairwise distance distributions between residues. "the distogram head predicts binned pairwise distance distributions from the final "
- DSSP: A standard algorithm for assigning secondary structure from protein coordinates. "Using DSSP~\citep{kabsch1983dictionary}, we identified internal loop regions in each target protein"
- Electrostatic complementarity: Favorable interactions between oppositely charged residues across strands or interfaces. "Electrostatic complementarity steering induces hairpin formation."
- ESM-2: A BERT-style protein LLM used to encode sequences into initial representations. "implemented as the BERT-style ESM-2~\cite{lin2023evolutionary}"
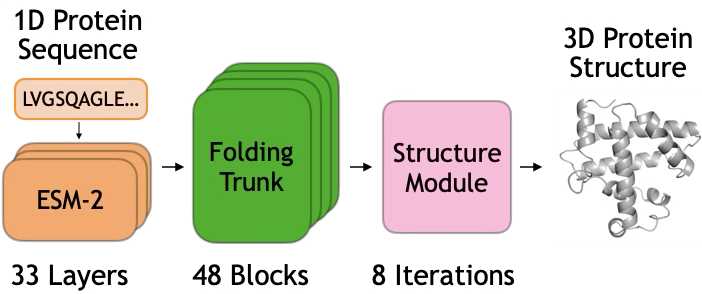
- ESMFold: A protein structure prediction model with a LLM encoder, folding trunk, and structure module. "We study these questions in ESMFold."
- Folding trunk: The core iterative module that refines sequence and pairwise representations en route to structure. "We identify two computational stages in the folding trunk."
- Helix-turn-helix motif: A structural motif of two helices connected by a short turn. "a target protein containing a helix-turn-helix motif."
- Hydrogen bond: A non-covalent interaction (here, backbone N–H to C=O) stabilizing secondary structures. "We measure hairpin induction by the percentage of cross-strand residue pairs forming backbone hydrogen bonds (N-O distance \AA)."
- Invariant point attention (IPA): A geometric attention mechanism invariant to rotations and translations, used in structure modules. "based on invariant point attention (IPA, ~\citet{jumper2021highly})"
- Learned positional embedding: Trainable vectors encoding relative/absolute position, used here to initialize pairwise representations. "The pair representation is initialized using a learned positional embedding."
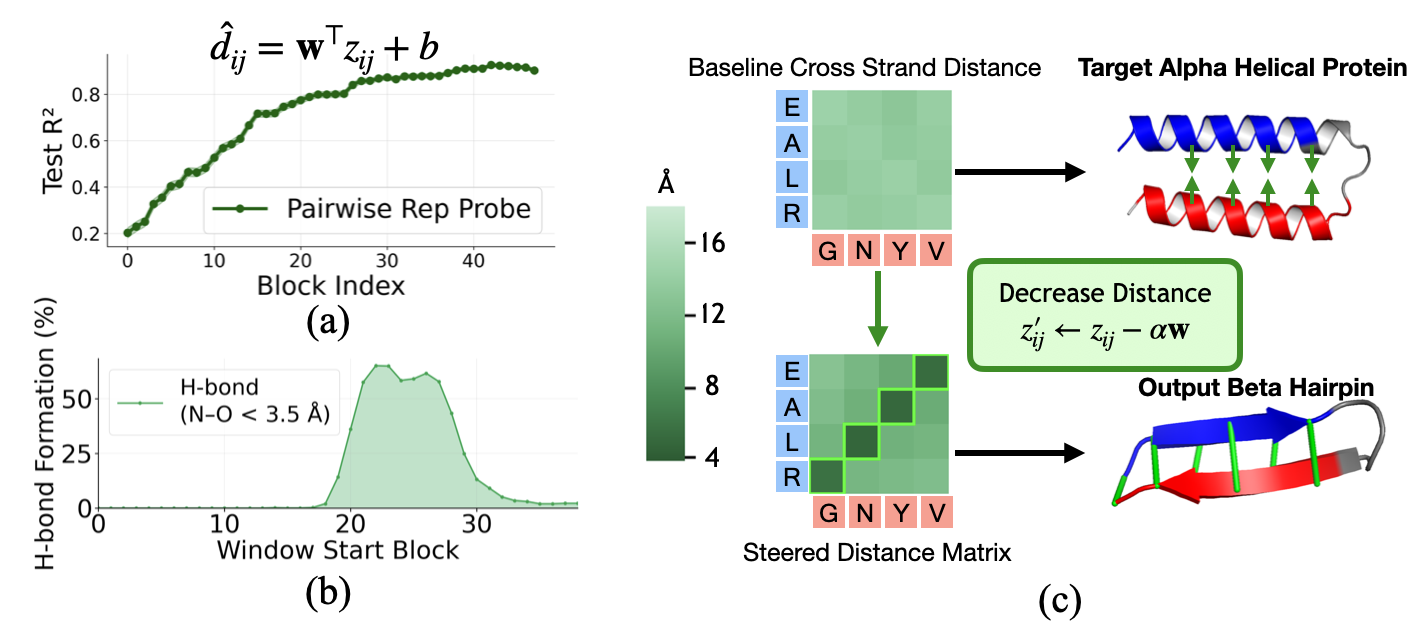
- Linear probe: A linear model trained to read out a property (e.g., distance) from internal representations. "We test this by training linear probes to predict pairwise C distance from at each block"
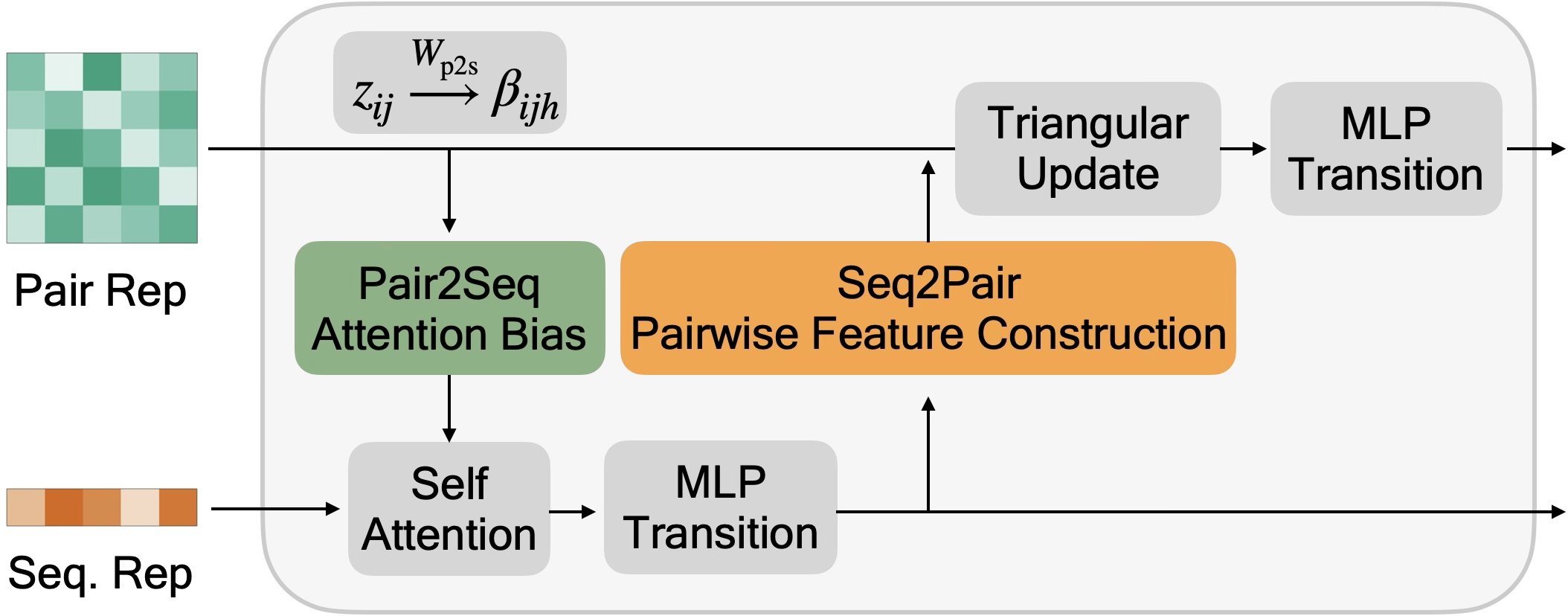
- Pair2seq (pair-to-sequence bias): The pathway that projects pairwise features into scalar biases that modulate sequence self-attention. "The pair2seq pathway projects into a scalar bias for each attention head and residue pair, which modulates sequence self-attention"
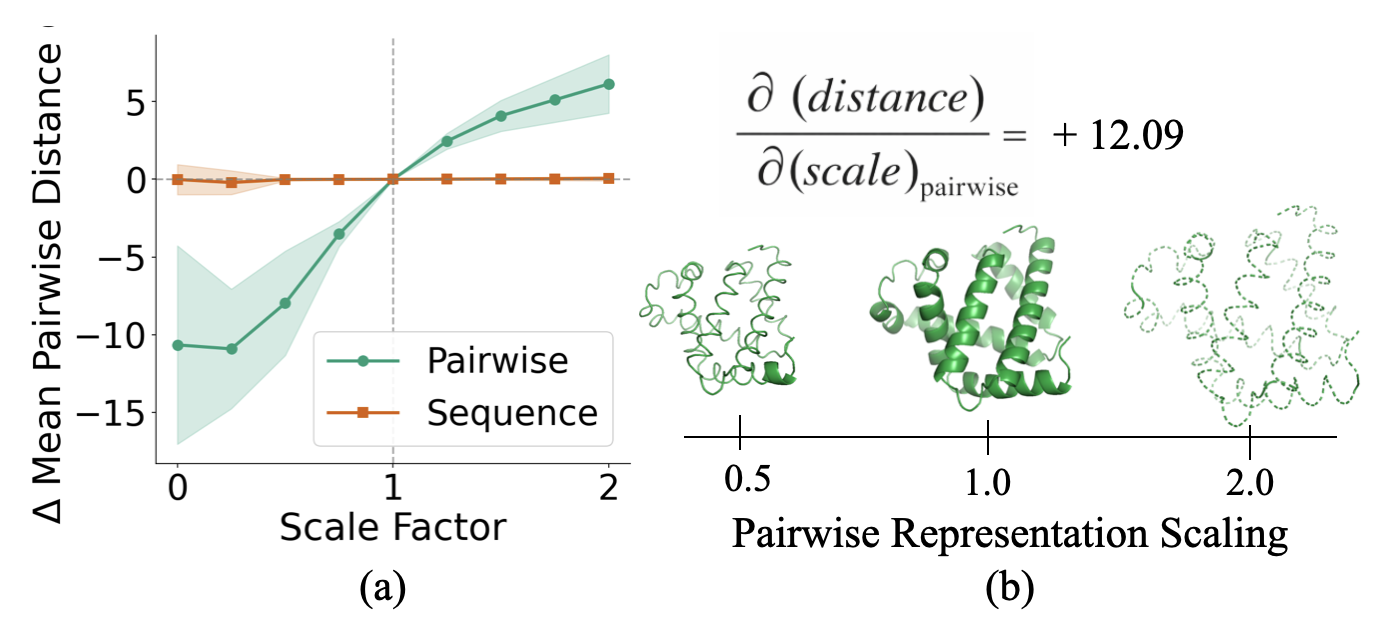
- Pairwise representation (z): A learned L×L×dz tensor encoding residue–residue relationships and geometry. "This suggests that the pairwise representation z has taken over as the primary carrier of folding-relevant information."
- Protein Data Bank (PDB): A public repository of experimentally determined protein structures. "For donor hairpins, we assembled a dataset of about 80,000 beta hairpins extracted from the Protein Data Bank (PDB; \citealt{berman2000protein})."
- Protein LLM: A neural model trained on protein sequences to learn residue-level representations. "A protein LLM (ESM-2) encodes an amino acid sequence into an initial sequence representation"
- Radius of gyration: A scalar summarizing the spatial extent/compactness of a structure. "we filter out cases where the radius of gyration drops below $0.9$ times the baseline value"
- Recycle (recycling): Reapplying the folding trunk multiple times to iteratively refine predictions. "The trunk is reapplied up to four times; each application is referred to as a recycle."
- Residue: An amino acid incorporated into a protein chain. "Proteins are linear chains of amino acids; once incorporated into a protein, each amino acid is called a residue."
- ROC-AUC: A performance metric measuring separability (Area Under the ROC Curve). "we compute whether the bias cleanly separates contacting residue pairs (C distance 8\AA) from non-contacts, measuring this with ROC-AUC"
- Salt bridge: An electrostatic interaction between oppositely charged side chains (e.g., Lys–Glu). "antiparallel beta strands are often stabilized by salt bridges between oppositely charged residues (e.g., lysine (K) and glutamate (E)) on facing positions"
- Secondary structure: Local structural motifs such as alpha helices and beta strands. "secondary structure refers to local motifs such as alpha helices and beta strands"
- Seq2pair (sequence-to-pair): The pathway that builds pairwise features from sequence features to update z. "The seq2pair pathway converts the sequence representation into a pairwise update via Eq.~\eqref{eq:seq2pair}."
- Sliding-window ablation: Disabling a component over consecutive blocks to assess causal contribution. "We perform sliding-window ablations, disabling seq2pair for consecutive blocks while patching sequence at block 0"
- Steering (representation steering): Adding a direction vector to latent representations to induce targeted changes. "we steer the sequence representation toward opposite charges on the two helical regions flanking the loop"
- Structure module: The component that converts refined representations into 3D coordinates. "The third is a structure module "
- Template conditioning: Injecting external structural templates into the model’s pairwise representation. "Template conditioning in AlphaFold2 and Boltz-2 inject information directly into "
- Triangular attention: A specialized attention mechanism over residue pairs used to refine pairwise features. "Second, is refined via triangular multiplicative updates and triangular attention~\cite{jumper2021highly}"
- Triangular multiplicative updates: Pairwise update operations combining information via triangle relations (i–k–j). "Second, is refined via triangular multiplicative updates and triangular attention~\cite{jumper2021highly}"
Collections
Sign up for free to add this paper to one or more collections.