Protein Language Model Embeddings Improve Generalization of Implicit Transfer Operators
Abstract: Molecular dynamics (MD) is a central computational tool in physics, chemistry, and biology, enabling quantitative prediction of experimental observables as expectations over high-dimensional molecular distributions such as Boltzmann distributions and transition densities. However, conventional MD is fundamentally limited by the high computational cost required to generate independent samples. Generative molecular dynamics (GenMD) has recently emerged as an alternative, learning surrogates of molecular distributions either from data or through interaction with energy models. While these methods enable efficient sampling, their transferability across molecular systems is often limited. In this work, we show that incorporating auxiliary sources of information can improve the data efficiency and generalization of transferable implicit transfer operators (TITO) for molecular dynamics. We find that coarse-grained TITO models are substantially more data-efficient than Boltzmann Emulators, and that incorporating protein LLM (pLM) embeddings further improves out-of-distribution generalization. Our approach, PLaTITO, achieves state-of-the-art performance on equilibrium sampling benchmarks for out-of-distribution protein systems, including fast-folding proteins. We further study the impact of additional conditioning signals -- such as structural embeddings, temperature, and large-language-model-derived embeddings -- on model performance.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
Overview
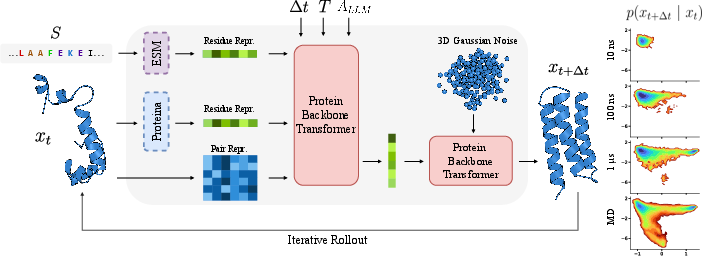
This paper is about making computer simulations of proteins much faster and smarter. Proteins move and change shape over time, and scientists often use heavy-duty simulations (called molecular dynamics, or MD) to watch this happen. The problem: MD is very slow and expensive to run. The authors build a new AI method, called PLaTITO, that learns to “skip ahead” in time and still predict realistic protein shapes. It uses extra “hints” from powerful protein LLMs (AI trained on millions of protein sequences) to work better on new, unseen proteins.
What questions did the researchers ask?
Before diving in, here are the main questions they wanted to answer:
- Can we train an AI to predict how a protein’s shape changes over longer time jumps (seconds in the real world) without simulating every tiny step (femtoseconds)?
- Can adding useful “hints” about the protein—like its sequence’s learned features from a protein LLM—make the AI more accurate and need less data?
- Will this AI generalize (work well) on proteins it never saw during training?
- Can it also capture how folding speeds change with temperature in a realistic way (not just following a simple rule)?
How did they do it?
The problem with regular simulations
- MD simulates motion in ultra-tiny time steps (think: moving one grain of sand at a time across a beach), but real protein motions happen at much longer times. Bridging that gap costs a ton of computer time.
Their shortcut: a “time jump” predictor
- Instead of stepping one tiny moment at a time, the model learns to answer: “Given the protein’s shape right now, what might it look like after a bigger jump in time?”
- This kind of model is called an implicit transfer operator (ITO). You can think of it like a weather forecast that predicts tomorrow’s weather in one shot, instead of simulating every minute of today’s weather.
- To train it, they use a technique called flow matching. In simple terms: teach a neural network to smoothly morph random noise into a realistic “future” protein shape. This makes sampling future shapes fast and efficient.
Adding helpful hints (embeddings)
They tried three kinds of extra inputs (embeddings) to help the model:
- Protein LLM embeddings (from ESM): These are features learned from billions of protein sequences. They capture useful patterns like which amino acids tend to go together and often relate to structure and function. This version is called PLaTITO.
- Structure embeddings (from Proteina): Features learned from lots of protein backbones that help the model understand 3D geometry.
- LLM-derived annotations: Hints from a general LLM that looked at protein metadata (like where in the cell the protein lives) and guessed whether simulating the protein alone makes sense.
They also built a larger version (PLaTITO-Big) with more model capacity and larger sequence embeddings.
What exactly does the model see?
- The model works on a simplified protein backbone (the “C-alpha” atoms), which keeps things fast.
- It’s conditioned on:
- the current protein shape,
- the amino acid sequence (plus embeddings),
- the temperature,
- and the chosen time jump length.
Training data and evaluation
- They trained on a big MD dataset of many different proteins at multiple temperatures (totaling about 56 milliseconds of MD time).
- They then tested on “out-of-distribution” proteins (new ones the model didn’t see), including “fast-folding proteins” that are commonly used as benchmarks.
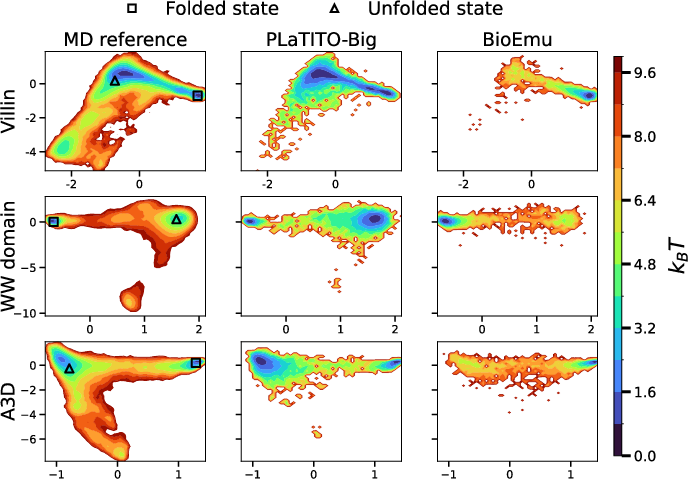
- They compared against a strong baseline called BioEmu, which is a different AI approach (a Boltzmann emulator) that focuses on reproducing the set of shapes a protein occupies at equilibrium (its most likely shapes over time).
What did they find?
Here are the key results and why they matter:
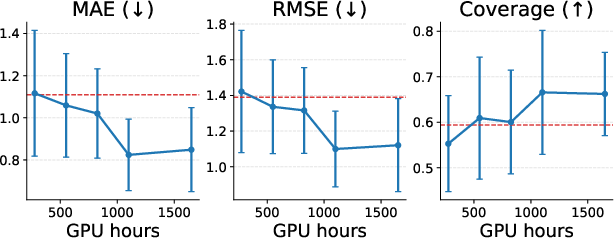
- Better generalization with less data:
- Adding protein LLM embeddings (PLaTITO) made the model more accurate than the basic version (TITO), even though both used the same training budget.
- The bigger model (PLaTITO-Big) achieved state-of-the-art results on equilibrium sampling for unseen proteins, beating BioEmu while using about 10× less compute and much less training data.
- Why it matters: Using “hints” from protein LLMs makes learning faster and transfers better to new proteins, which saves time and money.
- Accurate equilibrium behavior:
- The model reproduces the “free-energy landscape” of benchmark proteins—think of this as a map of the most likely shapes a protein can take, like hills and valleys where valleys are preferred shapes. PLaTITO-Big matched these maps very well.
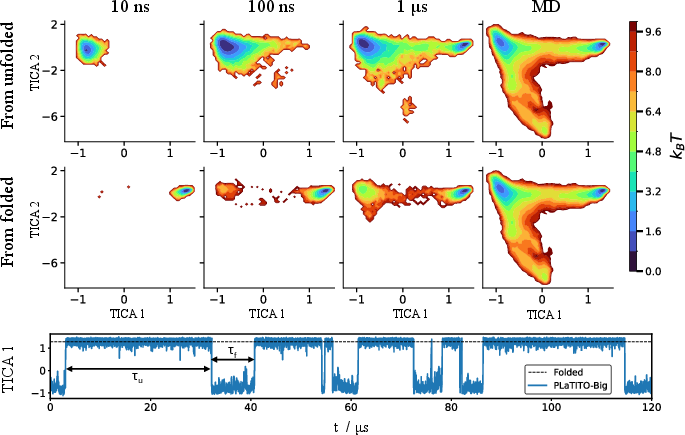
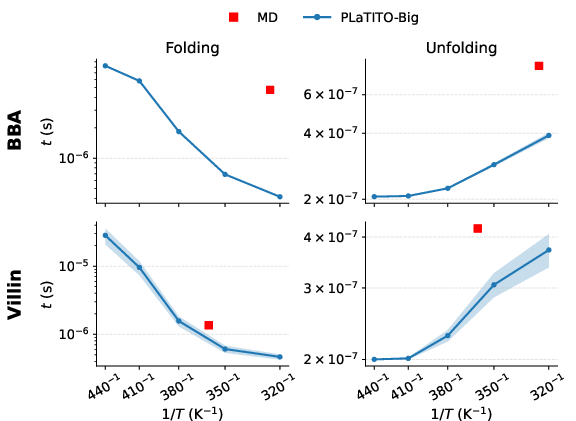
- Long-timescale dynamics:
- The model can roll out long trajectories that show proteins folding and unfolding multiple times, not just snapshotting static shapes.
- It tends to predict faster transitions than full MD (which the authors expected), but it still captures the overall behavior.
- Realistic temperature effects:
- Protein folding speeds do not always follow a simple “heat makes everything faster in the same way” rule (called Arrhenius behavior). The model recovers this more complex, “non-Arrhenius” pattern, which is what scientists see in real protein folding.
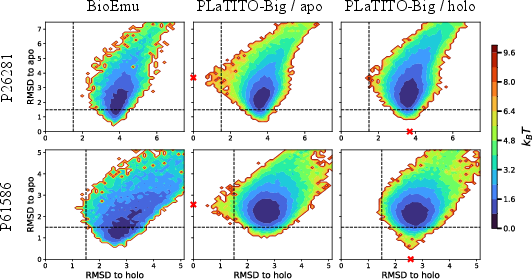
- Cryptic pockets (hard cases related to drug discovery):
- The model explores a wide range of shapes and sometimes reaches both “apo” (no ligand bound) and “holo” (ligand bound)–like shapes. It does better than the baseline in some tough cases, though there is still room to improve how clearly it separates long-lived states.
Why does this matter?
- Faster discovery: If an AI can accurately “jump ahead” in protein motion, scientists can explore protein behavior much faster than with classical simulations.
- Better generalization: Using protein LLMs as hints makes the method more robust on new proteins, which is crucial for real-world use (like studying diseases or designing drugs).
- Physical realism: Capturing realistic temperature effects and long-timescale folding/unfolding suggests the model learns meaningful dynamics, not just pretty static pictures.
- Drug design potential: Exploring rare shapes (like cryptic pockets) can help find new drug targets that are hard to spot otherwise.
Limitations to keep in mind
- Coarse-grained view: The model uses only the protein backbone (C-alpha atoms), not the full atom details. This is fast but can miss side-chain movements that matter for precise interactions.
- Not guaranteed exact physics: The method learns from data and doesn’t strictly enforce all the rules of physics. It sometimes underestimates how long processes take and can miss some stable states.
- Mixed results from extra hints: Structure embeddings helped a bit; language-model-derived metadata hints (from a general LLM) didn’t help in this setup.
Takeaway
PLaTITO is a fast, data-efficient way to predict how proteins move over time. By adding “hints” from protein LLMs, it generalizes well to new proteins and outperforms a strong baseline while using much less compute. It captures realistic behavior—like complex temperature effects—and shows promise for studying protein dynamics and helping drug discovery. With more data, larger models, and eventually full-atom detail, it could become a powerful tool for exploring the moving, living world of proteins.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a consolidated list of unresolved issues that the paper either acknowledges or leaves unexplored, formulated to guide concrete follow-up work:
- All-atom fidelity: The model operates on Cα-only coarse-grained representations; how to recover accurate side-chain dynamics, hydrogen bonding, sterics, and ligand-specific interactions via backmapping or joint CG–AA modeling remains unaddressed.
- Geometry/sterics constraints: No explicit enforcement of protein backbone geometry or clash-avoidance at the Cα level; methods to impose geometric priors or constraints during training/sampling are not explored.
- Formal dynamical guarantees: The approach lacks guarantees for detailed balance, stationarity under the target ensemble, semigroup self-consistency (Chapman–Kolmogorov), and long-horizon stability; metrics and loss terms to enforce or monitor these properties are not developed.
- Semigroup consistency across Δt: Although Δt is an input, the model is not constrained to produce self-consistent multi-step transitions (composition over shorter Δt vs one long Δt); tests and regularizers for semigroup consistency are absent.
- Long-horizon error accumulation: Stability and drift under very long iterative rollouts (beyond the presented horizons) are not quantified; strategies such as MH correction, periodic re-equilibration, or spectral regularization are not examined.
- Systematic timescale bias: The model underestimates MFPTs relative to MD; no calibration or correction schemes (e.g., spectral matching, rescaling based on implied timescales) are proposed.
- Temperature generalization: Non-Arrhenius trends are shown for a few proteins, but the model’s interpolation/extrapolation behavior across broader temperature ranges and different proteins is not systematically characterized.
- Conditioning beyond temperature: Effects of other thermodynamic/solution variables (pressure, ionic strength, pH, cofactor/ligand presence, crowding) are not modeled; extending conditioning to additional state variables is left open.
- Cross force-field robustness: Training and test trajectories arise from different force fields (e.g., mdCATH vs CHARMM22 fast-folders), but a systematic study of cross–force-field generalization is missing.
- Coverage of biomolecular scope: The model is trained on single-domain proteins up to 200 residues; transfer to larger proteins, multi-domain architectures, membrane proteins, intrinsically disordered proteins, and protein–protein or protein–nucleic acid complexes is untested.
- Cryptic pocket benchmarks: Only four cases are evaluated with Cα local RMSD; richer pocket metrics (volume, druggability, SASA changes) and ligand-aware assessments are not conducted.
- Kinetics validation breadth: Folding/unfolding kinetics are analyzed for a limited set; broader kinetic validation (implied timescales via MSM/VAMP on generated trajectories, transition path ensemble statistics, barrier heights) is not provided.
- Evaluation on projected spaces: Free-energy comparisons rely on TICA projections learned from MD; robustness to choice of features/lag times and evaluations in higher-dimensional spaces (e.g., contact maps, secondary structure, full distance matrices) are not examined.
- Equivariance vs non-equivariance: The chosen non-equivariant transformer is not compared to SE(3)-equivariant alternatives; the impact of equivariance on generalization, sample quality, and data efficiency remains unknown.
- pLM embedding design: The dependence on protein LLM choices (model size, layer selection, fine-tuning vs frozen, per-residue vs pooled features) and the mechanisms by which embeddings improve dynamics are not dissected via ablations.
- Structure embeddings overhead: Online computation of structure embeddings increases inference cost; benefits vs compute trade-offs, caching strategies, or learned in-model structural encoders are not explored.
- LLM-derived annotations: Conditioning on LLM annotations harms performance; alternative uses (data filtering, curriculum learning, sample weighting), improved prompting/metadata, or integrating uncertainty-aware treatment of annotations are not investigated.
- Data quality and biases: mdCATH trajectories may include non-native contexts (e.g., isolated domains missing partners); beyond an initial LLM heuristic, systematic detection/mitigation of dataset artifacts or selection biases is not assessed.
- Dataset size scaling laws: While compute scaling is shown, explicit data scaling laws (performance vs ms of MD) and minimal data requirements for transfer are not quantified.
- Baseline coverage: Direct empirical comparisons to other transfer-operator surrogates (e.g., Timewarp, (Equi)Jump/DeepJump) under identical datasets/splits are missing.
- Hyperparameter sensitivity: Effects of Δt choice, flow-matching schedules, noise distributions, and sampling hyperparameters on equilibrium and kinetic accuracy are not systematically ablated.
- Uncertainty quantification: The model provides point samples without calibrated uncertainty estimates; Bayesian/ensemble methods or stochasticity-aware diagnostics for transition density uncertainty are not provided.
- Ergodicity and mode coverage: Aside from TICA-coverage metrics, rigorous tests of ergodicity/mixing (e.g., state visitation over long horizons, recurrence times) are not presented; failure modes where metastable basins are not formed are noted but not systematically analyzed.
- Backmapping and downstream validation: Procedures to backmap CG trajectories to all-atom with solvent and to validate against experimental observables (e.g., SAXS, NMR, HDX) are not developed.
- Reproducibility constraints: Some evaluation data (fast-folders) are proprietary; complete reproducibility and community benchmarking are limited without fully open datasets and standardized pipelines.
- Computational efficiency at scale: Inference-time cost for very long trajectories and large proteins (including structure-embedding overhead) is not profiled; strategies for acceleration (distillation, pruning, caching) are unaddressed.
Practical Applications
Overview
Below we distill actionable, real-world applications that follow from the paper’s findings and innovations on PLaTITO (Protein-Language-aware Transferable Implicit Transfer Operators). We group applications into Immediate (deployable now) and Long-Term (requiring further R&D, scaling, or integration). Each item notes sectors, potential tools/workflows, and key assumptions or dependencies that impact feasibility.
Immediate Applications
The following can be piloted or adopted with the current PLaTITO/PLaTITO-Big capabilities (coarse-grained Cα models; conditioning on sequence, temperature, and time step; integration with pretrained pLMs; demonstrated generalization to unseen proteins; equilibrium/kinetics approximation; cryptic pocket exploration).
- Accelerated equilibrium ensemble generation for proteins
- Sector: healthcare/biotech (drug discovery), academia (structural biology)
- Tools/workflows:
- Use PLaTITO to rapidly generate microsecond-scale conformational ensembles from static structures or unfolded states; feed ensembles into docking and induced-fit scoring workflows
- Build low-dimensional free-energy surfaces (e.g., via TICA) to map folded/unfolded basins and intermediates for target selection
- Assumptions/dependencies: Cα-only coarse-grained representation (no side-chains); backmapping to all-atom needed for docking or chemistry-specific steps; accuracy best for systems similar to training distribution (single domains, up to ~200 residues)
- Cryptic pocket scouting and triage
- Sector: healthcare/biotech (hit discovery, allosteric targeting), academia (biophysics)
- Tools/workflows:
- Sample ensembles from apo and holo starting states; compute pocket-residue RMSDs or pocket-volume metrics to flag cryptic pocket opening tendencies
- Triage targets for in-depth MD or experimental follow-up (fragment soaking, covalent library screening)
- Assumptions/dependencies: Coarse-grained model may not capture fine side-chain gating; feasibility improved by pairing with fast backmapping and side-chain packing tools; energetic separation of metastable basins may be underestimated
- Warm-starts for enhanced sampling and MSM construction
- Sector: software/ML for computational chemistry; academia (simulation methods)
- Tools/workflows:
- Seed enhanced sampling (e.g., metadynamics, REFs) or short all-atom MD from PLaTITO-generated diverse conformers to improve coverage and reduce burn-in
- Initialize Markov State Models (MSMs) with broader state coverage from cheap PLaTITO rollouts, then refine transition probabilities with targeted MD
- Assumptions/dependencies: Underestimation of timescales (variational bias) should be considered; MSM construction requires consistent backmapping and careful validation
- Temperature-dependent kinetic hypothesis generation
- Sector: academia (biophysics), biotech (formulation/stability), education
- Tools/workflows:
- Rapidly assess folding/unfolding rate trends across temperatures to guide experiment design (e.g., define temperature windows for HDX-MS or DSC)
- Use non-Arrhenius trends captured by PLaTITO-Big as qualitative indicators of rugged free-energy landscapes
- Assumptions/dependencies: Quantitative kinetics may be faster than MD/experiment; results best used as qualitative guidance; generalization outside trained temperature range is uncertain
- Early-stage protein engineering triage for dynamic stability
- Sector: biotech (protein design), academia
- Tools/workflows:
- For sets of sequence variants, use PLaTITO (conditioned on sequence + pLM embeddings) to compare qualitative changes in folding basin populations or barrier-crossing frequency
- Rank-order variants for experimental testing to shift dynamic equilibria
- Assumptions/dependencies: Paper does not directly validate mutational scans; side-chain effects are not explicit; use as hypothesis generation, not final decision
- Interactive education and communication of protein dynamics
- Sector: education, outreach
- Tools/workflows:
- Classroom or web apps that visualize microsecond-scale folding/unfolding and basin exploration generated by PLaTITO at low compute cost
- Assumptions/dependencies: Coarse-grained trajectories; ensure clear communication of limitations vs. all-atom MD
- Cost and carbon footprint reduction in MD-centric projects
- Sector: policy (research funding, sustainability), industry R&D operations
- Tools/workflows:
- Replace large MD pre-runs with PLaTITO rollouts to reduce GPU hours for early exploration; use targeted MD only where needed
- Track compute and energy savings; incorporate into lab or enterprise “green computing” metrics
- Assumptions/dependencies: Requires institutional acceptance of surrogate modeling as a pre-screen; downstream validation remains necessary
- Software integration modules and APIs
- Sector: software/tools
- Tools/workflows:
- Provide a PLaTITO Python library and CLI with adapters for MDAnalysis/MDTraj, OpenMM, and docking suites (e.g., induced-fit pipelines)
- Offer a “Conformational Ensemble Service (CES)” API: upload sequence + structure, return Cα ensemble + TICA projections + suggested seeds for MD
- Assumptions/dependencies: Precomputed pLM embeddings (e.g., ESM) advisable; proper licensing for datasets/models; provide standardized backmapping support
- Benchmarking for GenMD research
- Sector: academia (ML for physical sciences)
- Tools/workflows:
- Use PLaTITO as a baseline for equilibrium sampling and transfer-learning across protein families; study the effect of auxiliary conditioning (pLM/structural embeddings)
- Assumptions/dependencies: Public access to mdCATH-like datasets and trained checkpoints; consistent evaluation protocols (e.g., TICA settings)
Long-Term Applications
These depend on additional research, scaling, and/or integration with all-atom physics, priors, or expanded training data (complexes, membranes, ligands). They outline products and workflows that PLaTITO enables with maturation.
- Dynamics-informed drug discovery at scale (Dynamics-as-a-Service)
- Sector: healthcare/biotech, pharma platforms
- Tools/workflows:
- Cloud service that generates dynamic ensembles across proteomes; feeds docking/induced-fit workflows; prioritizes cryptic-pocket-ready targets; triggers targeted MD/FEP on selected conformers
- Active-learning loops coupling medicinal chemistry and surrogate dynamics to enrich chemotypes for allosteric or state-specific binding
- Assumptions/dependencies: All-atom backmapping and side-chain dynamics critical; calibration to specific force fields; validated links between coarse-grained ensembles and binding outcomes
- All-atom extension with Boltzmann priors and physical constraints
- Sector: academia, software/tools, pharma
- Tools/workflows:
- Extend to side-chains and solvent or tightly couple to backmapping plus energy-based filters; integrate Boltzmann-prior regularization to improve equilibrium fidelity and detailed balance; add self-consistency checks (e.g., Chapman–Kolmogorov)
- Assumptions/dependencies: Training on larger, diverse, high-quality MD datasets; computational scaling; careful validation against kinetics and thermodynamics benchmarks
- Protein–ligand and protein–protein complex dynamics
- Sector: drug discovery (allosteric modulators, PPI inhibitors), structural biology
- Tools/workflows:
- Condition on ligand identity/pose and partner sequences to model induced-fit dynamics; rapidly explore binding/unbinding pathways and transient pockets in complexes or membranes
- Assumptions/dependencies: New training data for complexes/ligands; representations that fuse ligand/protein features; validation against experimental kinetics
- Design of dynamic proteins and switchable systems
- Sector: protein engineering, synthetic biology
- Tools/workflows:
- Optimize sequences for target dynamic landscapes (e.g., two-state switches, preorganized loops) by coupling PLaTITO with sequence design models and Bayesian optimization
- Assumptions/dependencies: Requires differentiable or sample-efficient objective functions mapping dynamics to fitness; side-chain and functional readouts needed
- Materials and polymer dynamics beyond proteins
- Sector: materials science, energy, soft matter
- Tools/workflows:
- Adapt the ITO + language-embedding paradigm to polymer chains or soft materials (e.g., sequence-aware polymer descriptors) to accelerate morphology and transport predictions
- Assumptions/dependencies: Domain-specific sequence/structure embeddings; curated MD datasets; validation on diffusion/viscoelastic observables
- Experimental co-design and high-throughput planning
- Sector: academia, biotech
- Tools/workflows:
- Integrate surrogate dynamics with Bayesian experimental design to plan NMR/HDX-MS/smFRET experiments targeting slow modes or state populations; iteratively refine the model with sparse experimental constraints
- Assumptions/dependencies: Interfaces that assimilate experimental data; protocols for uncertainty quantification; standardized data exchange formats
- Standards, validation, and governance for AI surrogates in MD
- Sector: policy, research governance, industry standards
- Tools/workflows:
- Establish benchmarking suites, confidence metrics, and reporting norms for AI-driven MD surrogates (equilibrium, kinetics, transferability); promote open MD datasets and compute-carbon accounting guidelines
- Assumptions/dependencies: Community consensus and funding; alignment with regulatory expectations for in silico evidence in R&D
- Integrated enterprise platforms with E2E pipelines
- Sector: software/tools for pharma and biotech
- Tools/workflows:
- Productize an end-to-end pipeline that takes sequences/structures, performs dynamics-aware triage, runs targeted MD/FEP, and manages data/compute provenance for decision-making
- Assumptions/dependencies: Robust MLOps, data governance, and IP/licensing for pretrained models and MD datasets
Key Cross-Cutting Dependencies and Assumptions
- Representation limits: Current models are Cα-only and non-equivariant; side-chain rearrangements and explicit solvent effects are absent.
- Physical guarantees: No formal guarantees of unbiased dynamics, detailed balance, or semigroup properties; timescales tend to be underestimated.
- Training distribution: Generalization is best for systems similar to training data (single domains ≤~200 residues, isolated from binding partners). Performance on complexes, membranes, PTMs remains unproven.
- Embedding availability: pLM embeddings (e.g., ESM Cambrian) are pivotal; ensure licensing and access. Structure-embedding gains are modest; LLM-derived metadata did not help in this study.
- Compute/data: While more efficient than Boltzmann emulators, training still requires substantial GPU hours and curated MD datasets (e.g., mdCATH); inference is comparatively cheap.
- Backmapping: Many downstream applications need reliable, fast, and physically plausible backmapping to all-atom representation, plus optional side-chain sampling and local relaxation.
Glossary
- Activation energy: The energy barrier parameter that influences temperature-dependent reaction rates in Arrhenius kinetics. "idealized exponential temperature dependence in the activation energy described by \citeauthor{arrhenius1889reaktionsgeschwindigkeit}, $k(T) = A \exp \left(-E_a/k_B T\right),"</li> <li><strong>AFDB</strong>: The AlphaFold Database of predicted protein structures used to scale training with synthetic backbones. "by scaling training to synthetic structures from AFDB \cite{varadi2022alphafold}."</li> <li><strong>Amino-acid sequence</strong>: The ordered string of residues that defines a protein’s primary structure. "defined by backbone coordinates $x_tST$,"</li> <li><strong>Arrhenius</strong>: A kinetic model predicting exponential temperature dependence of reaction rates; deviations are termed non-Arrhenius. "We show that the temperature-dependent kinetics learned by PLaTITO are non-Arrhenius,"</li> <li><strong>Attention biases</strong>: Additive terms in transformer attention that condition interactions between residues. "where pair representations are used as attention biases."</li> <li><strong>Apo state</strong>: The ligand-free conformation of a protein. "initialized either from the {\it apo} or from the {\it holo} state"</li> <li><strong>Backbone coordinates</strong>: The 3D positions of main-chain atoms (often Cα) describing protein geometry. "defined by backbone coordinates $x_t$"</li> <li><strong>Binding affinities</strong>: Quantitative measures of interaction strength between molecules. "for stationary properties (e.g., binding affinities)"</li> <li><strong>Binding pocket residues</strong>: The subset of residues forming the ligand-binding site. "we compute local $C_\alpha$ RMSDs, using only binding pocket residues to the reference {\it apo} and {\it holo} structures."</li> <li><strong>Boltzmann constant</strong>: Physical constant relating temperature to energy scale in statistical mechanics. "where $p_iik_BT$ is the simulation temperature."</li> <li><strong>Boltzmann distribution</strong>: The equilibrium probability distribution over molecular states proportional to $\exp(-\beta U)$. "admits the Boltzmann distribution, $\mu(x) \propto \exp(-\beta U(x))$, as its invariant measure."</li> <li><strong>Boltzmann Emulators</strong>: Models that approximate equilibrium densities without strict theoretical guarantees, emphasizing scalability and validation. "Boltzmann Emulators (BE) \cite{NEURIPS2022_994545b2,lewis2025scalable,diez2024generation,jamun} sacrifice details and theoretical guarantees and instead focus on scaling and external validation."</li> <li><strong>Boltzmann Generators</strong>: Learned surrogates of the Boltzmann distribution that permit exact reweighting to the target density. "Boltzmann Generators (BG) are surrogates of the Boltzmann distribution, $\mu(x)$, learned from data, potential energy model or a combination of the two, and allow for exact reweighing to target Boltzmann density \cite{No2019}."</li> <li><strong>Cα backbone coordinates</strong>: Coarse-grained representation using alpha-carbon positions per residue. "using only the $C_{\alpha}x_t \in {\mathbb{R}^{3L}$"</li> <li><strong>Chapman-Kolmogorov</strong>: The semigroup consistency property required for Markov processes across time scales. "semi-group self-consistency (Chapman-Kolmogorov),"</li> <li><strong>Coarse-grained representation</strong>: A simplified model that reduces atomic detail to lower computational cost (e.g., Cα-only). "we adopt a coarse-grained representation of the proteins using only the $C_{\alpha}$ backbone coordinates"</li> <li><strong>Conditional Flow Matching</strong>: Training scheme that regresses a neural velocity field to analytic conditional paths. "we adopt Conditional Flow Matching (CFM) that constructs conditional probability paths $p_s(z \mid z_1)$"
- Cryptic binding pockets: Rare, low-population protein conformations revealing hidden ligand-binding sites. "cryptic binding pockets: lowly populated conformational states of proteins which might be targeted by therapeutics"
- DeepJump: A generative approach using stochastic interpolants to model long-time transitions without latent Gaussians. "later, the authors extended this work in DeepJump \cite{costa2025accelerating} training on the larger mdCATH dataset \cite{mirarchi2024mdcath} illustrating limited transferability to the fast-folders."
- Diffusion models: Generative models that learn distributions via stochastic processes; used here for conditional transitions. "ITO \cite{schreiner2023implicit} introduced the use of conditional diffusion models to build multiple time-scale MD surrogate models"
- Dynamic graphical models: Probabilistic models capturing temporal dependencies via graph structures. "dynamic graphical models \cite{olsson2019dynamic,Hempel2022}"
- Equilibrium distribution: The stationary molecular distribution achieved after sufficient mixing time. "We first evaluate the ability of our models to reproduce the equilibrium distribution of the fast-folding proteins \cite{lindorff2011fast}."
- Fast-folding proteins: Small proteins with microsecond-scale folding kinetics used as benchmarks. "including fast-folding proteins."
- Femtosecond-scale time-steps: Extremely small integration steps used in explicit MD for numerical stability. "Classical MD relies on explicit numerical integration with femtosecond-scale time-steps,"
- Flow map: The transformation solving an ODE that pushes a base distribution along a density path. "The flow map is defined as the solution of an ordinary differential equation (ODE)"
- Flow-matching: A generative framework that learns a velocity field to transport a base to a target distribution. "Flow-matching \cite{albergo2023building, liu2023flow, lipman2023flow} is a generative modeling framework"
- Force field: Parameterized potential energy model defining interatomic forces in simulations. "either from trajectory data or by learning from a potential energy model---or force field."
- Free energy landscape: The energy surface over conformational coordinates, whose basins correspond to metastable states. "consistent with complex rugged folding free energy landscapes"
- Generative molecular dynamics (GenMD): Methods that learn to sample molecular states without explicit time integration. "generative molecular dynamics (GenMD) methods have emerged \cite{Olsson2026},"
- Holo state: The ligand-bound conformation of a protein. "initialized either from the {\it apo} or from the {\it holo} state"
- Implicit Transfer Operator (ITO): A framework learning long-timestep transition densities directly from MD data. "Implicit Transfer Operator (ITO) learning \cite{schreiner2023implicit} provides a framework for modeling long-timestep molecular dynamics"
- Invariant measure: A distribution unchanged by the dynamics, such as the Boltzmann distribution under certain conditions. "as its invariant measure."
- Iterative roll-outs: Repeated application of learned transition models to simulate long trajectories. "perform 1,000 iterative roll-outs with a physical time step "
- Lag time: The time delay used in time-lagged analyses (e.g., TICA) to capture slow dynamics. "with a lag time of 10 ns."
- Langevin equation: A stochastic differential equation describing dynamics under thermal noise and potential forces. "by numerically integrating the Langevin equation \cite{langevin1908theorie} under a potential energy ."
- Latent flow variable: The interpolant bridging noise and data along a learned conditional path. "We introduce a latent flow variable ,"
- LLM-derived annotations: Heuristic labels and confidence scores extracted via LLMs to assess simulation suitability. "and LLM-derived annotations ."
- Long-timescale transition densities: Probability distributions over future states at large time steps. "most notably Boltzmann equilibrium distributions and long-timescale transition densities."
- Markov process: A memoryless stochastic process where the next state depends only on the current state. "This generates a discrete-time Markov process with transition density "
- Markov state models (MSMs): Discrete-state models estimating the transfer operator from time series. "including Markov state models (MSMs) \cite{prinz2011markov}"
- Mean first-passage times (MFPTs): Expected times for first transitions between states (e.g., folding/unfolding). "estimate folding and unfolding rates using mean first-passage times (MFPTs) following \citeauthor{schreiner2023implicit}"
- Metropolis-Hastings: A Monte Carlo correction ensuring unbiased sampling from a target distribution. "provides unbiased equilibrium samples through a Metropolis-Hastings correction"
- Molecular dynamics (MD): Simulation technique evolving molecular systems via numerical integration of physical laws. "Molecular dynamics (MD) simulations provide a computational bridge from microscopic physical laws to molecular phenomena,"
- Observable operator models: Operator-based models capturing time series dynamics via observable functions. "observable operator models \cite{Wu2015},"
- Off-equilibrium MD trajectories: Simulation data not sampled from equilibrium, used for training transfer models. "trained from scratch on diverse off-equilibrium MD trajectories across multiple temperatures"
- Ordinary differential equation (ODE): A differential equation governing the flow map in continuous transport. "an ordinary differential equation (ODE)"
- Potential energy: Scalar function U(x) defining energetic cost of configurations and driving forces. "under a potential energy ."
- Protein LLMs (pLMs): Sequence-trained models capturing structural and functional information in embeddings. "Protein LLMs (pLMs), trained on billions of unlabeled protein sequences,"
- Proteina: A structure-aware transformer architecture used for backbone-based representations. "Both and are non-equivariant transformer architectures based on Proteina \cite{geffner2025proteina}."
- Push-forward operator: The operator mapping a distribution through a function (flow map) to a new distribution. "where denotes the push-forward operator."
- Rectified flow: A linear conditional path formulation yielding constant velocity fields for training. "described in the rectified flow formulation \cite{liu2023flow}, ,"
- RMSD (Root-Mean-Square Deviation): A measure of structural difference between conformations. "we compute local RMSDs, using only binding pocket residues"
- Semi-group self-consistency: The requirement that multi-step dynamics compose consistently across time (Chapman-Kolmogorov). "semi-group self-consistency (Chapman-Kolmogorov),"
- Stationary distribution: The time-invariant distribution reached by the dynamics. "To approximate the stationary distribution, we keep only the final of each trajectory"
- Structure embeddings: Learned per-residue features from backbone geometry used as conditioning signals. "Incorporating structure embeddings (PLaTITO+Struct) provides a modest but consistent improvements across all metrics"
- Surrogate models: Learned approximations replacing expensive simulations to sample from target distributions. "Generative molecular dynamics (GenMD) has recently emerged as an alternative, learning surrogates of molecular distributions either from data or through interaction with energy models."
- Thermodynamic state: The macroscopic condition (e.g., temperature) defining equilibrium properties. "transfer across chemical space \cite{transbgs,tan2025scalable,akhound-sadegh2025progressive} and thermodynamic state \cite{moqvist2025thermodynamic, Schebek2024}."
- Time-lagged Independent Component Analysis (TICA): A dimensionality-reduction method extracting slow coordinates from time series. "we estimate a Time-lagged Independent Component Analysis (TICA) model"
- Timewarp: A flow-based generative model for MD transitions corrected via Metropolis-Hastings. "Timewarp \cite{klein2023timewarp} is a flow-based model that provides unbiased equilibrium samples through a Metropolis-Hastings correction"
- Transfer operator: The linear operator propagating probability densities over time. "Approximating molecular transition densities using transfer operator-based approaches has been widely studied"
- Transition density: The conditional probability of future states given current states over a time step. "transition density "
- Variational Approach for Markov Processes (VAMP): A variational method for learning slow dynamics and state decompositions. "through the variational approach for Markov processes (VAMP) \cite{Wu2019}."
- Variational principle: A bound indicating learned dynamics typically underestimate true time scales. "as a variational principle suggests that imperfect approximations of MD systematically underestimate time-scales \cite{Nske2014}."
- Velocity field: The vector field driving the flow that transports probability mass along the path. "where denotes the time-dependent velocity field"
- VAMPnets: Neural architectures that learn state memberships via the VAMP objective. "and VAMPnets \cite{mardt2018vampnets}."
Collections
Sign up for free to add this paper to one or more collections.