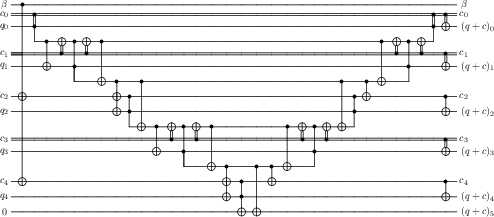
- The paper presents a quantum algorithm that efficiently block-encodes the full pre-Born-Oppenheimer Hamiltonian using a swap network and an alternating sign LCU.
- The methodology leverages a real-space grid with spectral shifting to accurately simulate coupled electron-nuclear dynamics while lowering resource overhead.
- The approach achieves an order-of-magnitude reduction in cost, paving the way for practical quantum simulations in photochemical and catalytic processes.
Efficient Simulation of Pre-Born-Oppenheimer Dynamics on Quantum Computers: Technical Overview
Introduction and Motivation
The simulation of quantum molecular dynamics beyond the Born-Oppenheimer approximation represents a crucial frontier, especially for photochemical and non-adiabatic processes where electron-nuclear correlations are non-negligible. Accurate classical simulations are plagued by exponential scaling, and traditionally, quantum algorithms have focused on electronic structure within the Born-Oppenheimer regime, neglecting direct simulation of coupled electron-nuclear dynamics. This paper introduces a quantum algorithm for the first-principles simulation of electron-nuclear dynamics using a first-quantized real-space grid representation. The method achieves significant efficiency improvements in the block-encoding of the full pre-Born-Oppenheimer molecular Hamiltonian, directly addressing several computational bottlenecks of prior approaches.
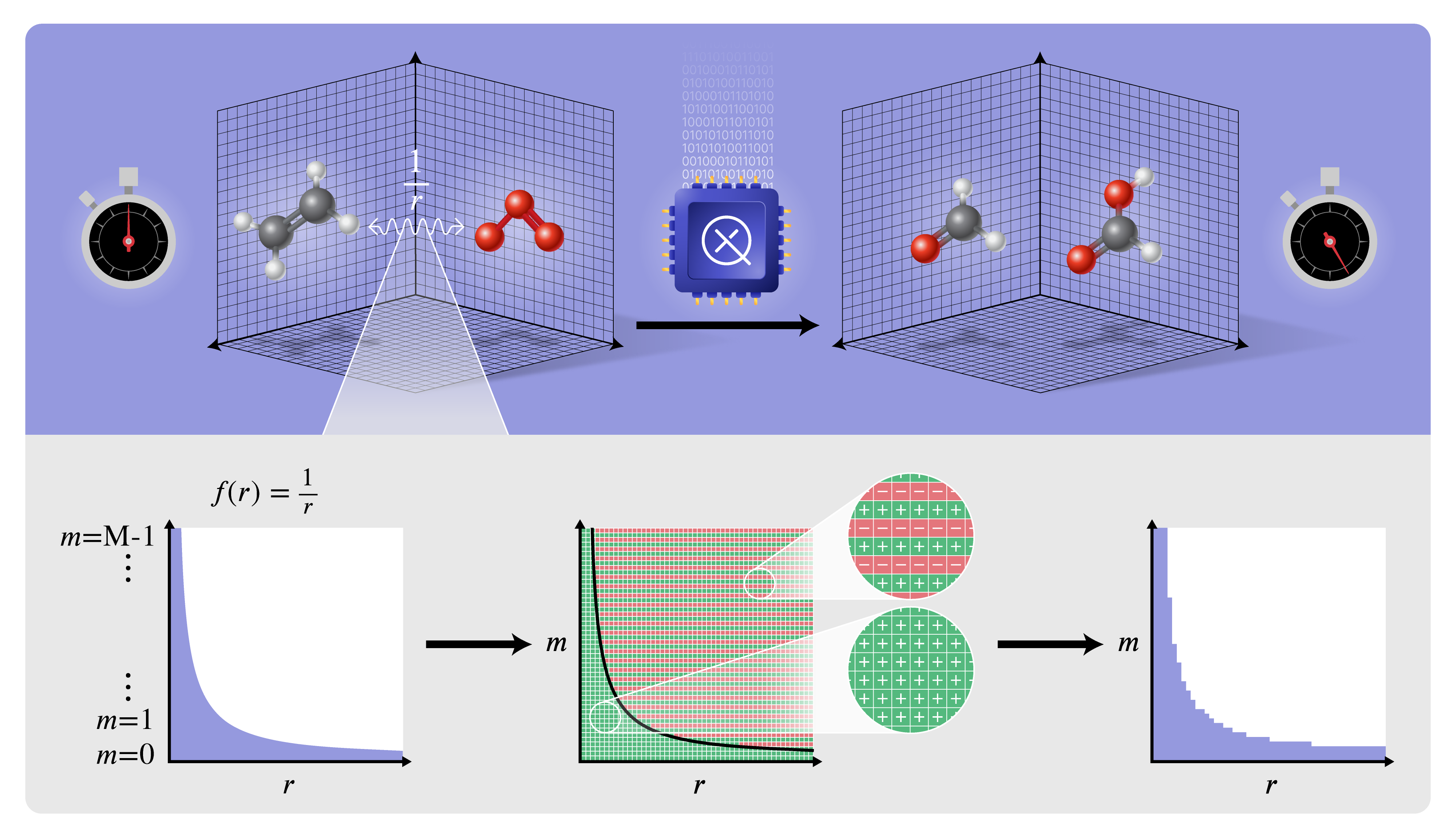
Figure 1: Quantum simulation of chemical reactions on a real-space grid, leveraging an LCU for the Coulomb $1/r$ interaction via an alternating sign technique efficiently implementable with quantum arithmetic.
Hamiltonian and Real-Space Encoding
The core physical model is the non-relativistic pre-Born-Oppenheimer Coulomb Hamiltonian:
H=i=1∑η2mi−∇i2+i=j∑(−1)σi+σj2rijζiζj
where η is the total particle number (electrons + nuclei), and rij are real-space separations. Both electrons and nuclei are discretized on a Cartesian ng-bit grid per coordinate. Care is taken with grid spacing (Δ), ensuring the resolution is sufficient so that configurations where two particles overlap have negligible weight in the dynamics. The wavefunction is realized as a tensor product over these grid registers, supporting efficient QFTs for kinetic energy evaluation.
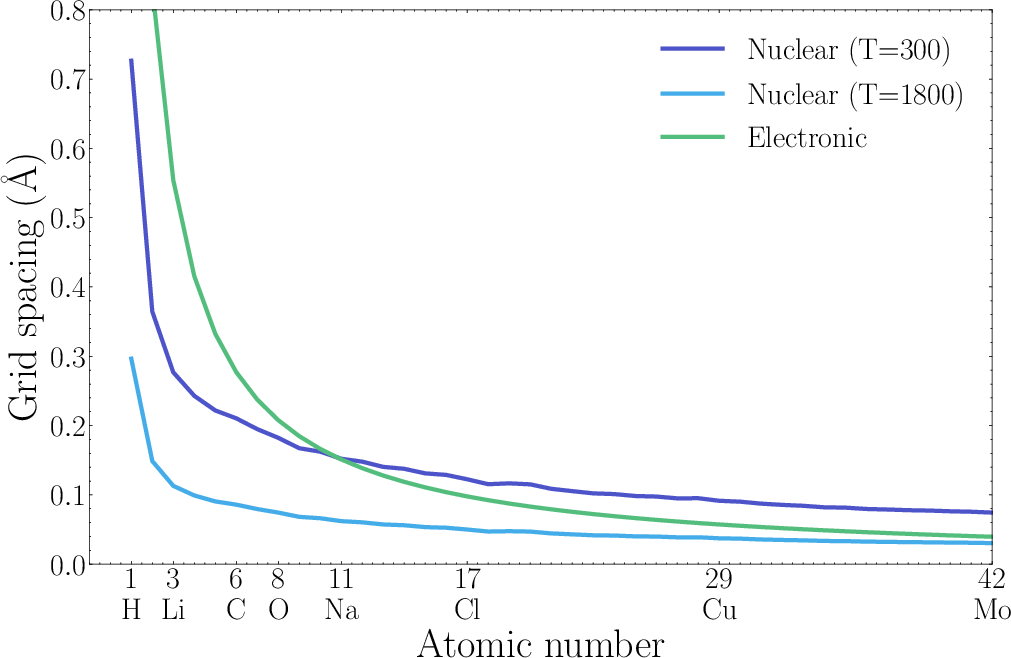
Figure 2: Grid spacings for different nuclei, calibrated for room temperature using de Broglie considerations.
Linear Scaling Block-Encoding Architecture
Previous quantum algorithms for such dynamics scale poorly, with a quadratic cost in the number of particles due to the O(η2) two-body interactions. This work introduces a "swap network" block-encoding scheme which generalizes the swap-based "swap up" method and enables block-encoding both kinetic and potential operators with linear resource scaling in η. This is achieved by decomposing interaction evaluation into a sequence of multiplexed swaps, enabling recursive tensor product application while avoiding redundant computation.
The potential operator V is efficiently encoded using an LCU decomposition over all particle pairs. This is realized by preparing registers encoding each pair, performing appropriate swaps, and block-encoding the Coulomb interaction via custom quantum arithmetic.
Alternating Sign Implementation for $1/r$ Block-Encoding
A central technical innovation is the alternating sign LCU implementation for the $1/r$ Coulomb potential:
The flexibility of the routine supports variable saturation for the potential (to account for physically relevant minimal nuclear separations) and a shifting technique to halve the 1-norm, both required for realistic systems.
Cost Analysis and Profiling
The resulting block-encoding of the Hamiltonian achieves Toffoli complexity:
O~(η+log2(1/ϵ))
per block-encoding, where ϵ is the target error, and the complexity for time-evolution of duration t is:
O~(η3t+ηlog(1/ϵ)).
This demonstrates strong polynomial scaling, reducing cost by more than an order of magnitude compared to previous approaches based, for example, on plane-wave bases or less optimized LCU/hybrid implementations. Resource estimates are provided for several reactions relevant to catalysis, atmospheric, and combustion chemistry.
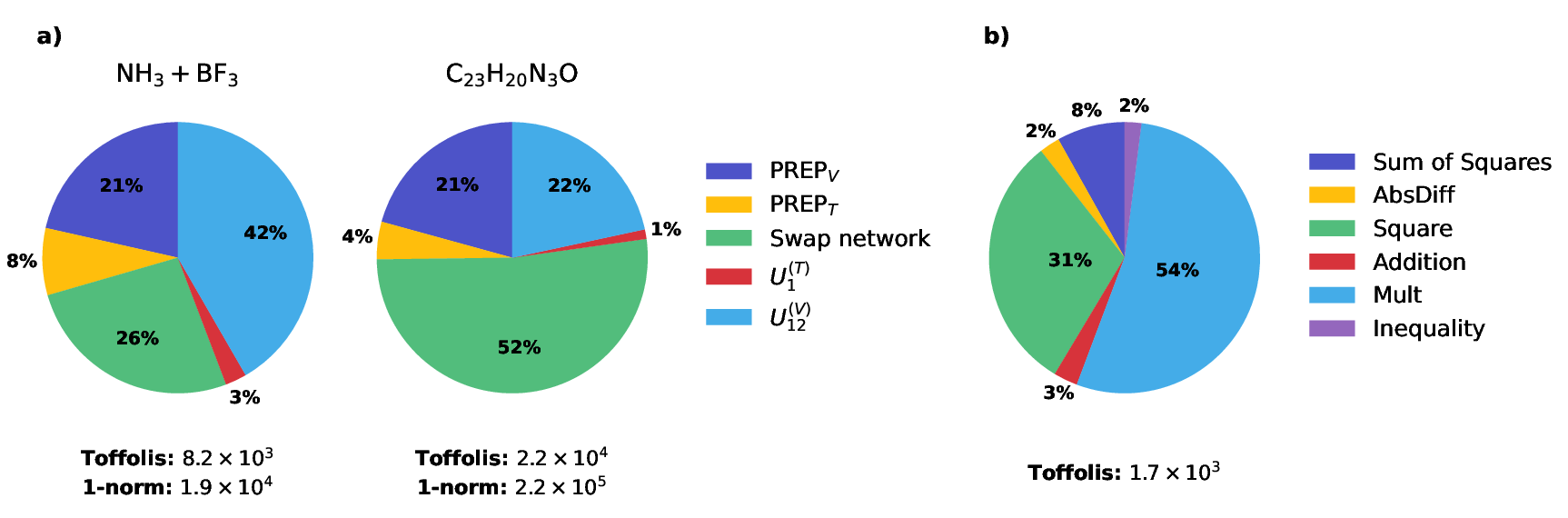
Figure 4: (a) Toffoli cost decomposition per walk operator for various chemical reactions; (b) Toffoli cost profile for the U12(V) implementation of the $1/r$ potential for the NH3+BF3 reaction.
The dominant cost at small system sizes arises from the alternating sign implementation of $1/r$, while for larger systems, the swap network cost (linear in η) dominates. Ancilla requirements remain subdominant—less than 26% of total qubits, even for large molecules.
Physically Motivated and Spectral Optimizations
- Variable Nuclear Saturation: Given that nuclear–nuclear pairs rarely reach separations less than a thermal or zero-point quantum estimate, the block-encoding only requires saturation of the $1/r$ divergence above a certain threshold, further reducing the 1-norm and consequently the gate cost.
- Spectral Shifting: Shifting the spectrum of the block-encoded operator (so [0,1/Δ]→[−1/2Δ,1/2Δ]) halves the necessary LCU coefficients, achieving an additional constant-factor gain with only a trivial energy shift.
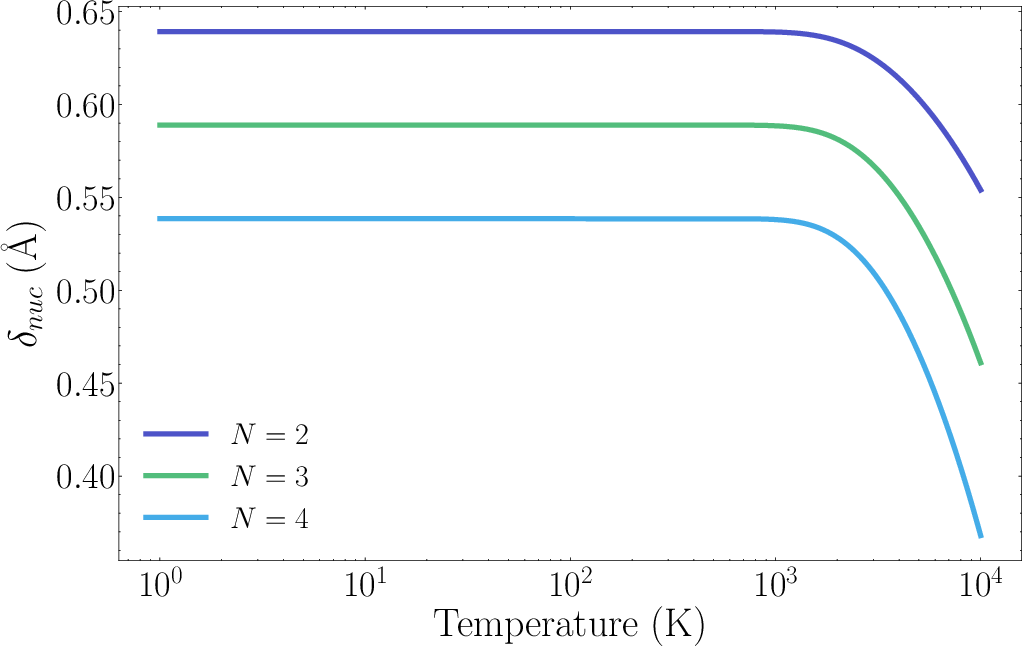
Figure 5: Minimum nuclear separation for H2 as a function of temperature, guiding the choice of saturation thresholds in the block-encoding.
Practical and Theoretical Implications
This algorithmic advance significantly improves the prospects for direct, black-box, pre-Born-Oppenheimer quantum simulation on early fault-tolerant quantum hardware, with closest short-term applications in:
- Photochemical mechanisms: Non-adiabatic processes central to light harvesting, photocatalysis, and atmospheric chemistry are now within resource reach for medium-sized molecules.
- Radical and proton-coupled electron transfer (PCET) chemistry: The approach handles electron–nuclear coupling without uncontrolled approximations, crucial for energy and catalytic applications.
- Mechanism-agnostic modeling: Because no electronic structure-based parametrization is required, the algorithm can serve in workflow automation for reaction screening where accurate pathways are not known a priori.
In the algorithmic domain, the primitives developed (linear-scaling swap networks, alternating sign LCU, modular block-encoding, and spectral shifting) have direct applicability to other many-body first-quantized quantum simulation problems and could inform future hardware-level optimizations.
Future Directions
- Pseudopotentials: Introducing effective core potentials could alleviate both the qubit and gate requirements further, crucial for larger systems and for incorporating relativistic effects.
- Thermodynamic environments and solvent effects: The real-space formalism can be adapted, as outlined in recent work, for coupled quantum-classical or open-system simulations.
- Further reduction of 1-norms and improved state preparation: Continuing innovation on LCU construction and initial state loading will remain areas for advancing practical quantum simulation.
Conclusion
This work provides the most efficient quantum algorithm to date for direct, first-principles simulation of coupled electron-nuclear dynamics in chemistry, attaining both strong theoretical guarantees and practical resource reductions. Its modular block-encoding framework, alternating sign LCU for potentials, and physical optimizations yield immediate gains and establish a blueprint for future quantum simulation algorithms targeting chemically and industrially relevant problems.