Enhanced Diffusion Sampling: Efficient Rare Event Sampling and Free Energy Calculation with Diffusion Models
Abstract: The rare-event sampling problem has long been the central limiting factor in molecular dynamics (MD), especially in biomolecular simulation. Recently, diffusion models such as BioEmu have emerged as powerful equilibrium samplers that generate independent samples from complex molecular distributions, eliminating the cost of sampling rare transition events. However, a sampling problem remains when computing observables that rely on states which are rare in equilibrium, for example folding free energies. Here, we introduce enhanced diffusion sampling, enabling efficient exploration of rare-event regions while preserving unbiased thermodynamic estimators. The key idea is to perform quantitatively accurate steering protocols to generate biased ensembles and subsequently recover equilibrium statistics via exact reweighting. We instantiate our framework in three algorithms: UmbrellaDiff (umbrella sampling with diffusion models), $Δ$G-Diff (free-energy differences via tilted ensembles), and MetaDiff (a batchwise analogue for metadynamics). Across toy systems, protein folding landscapes and folding free energies, our methods achieve fast, accurate, and scalable estimation of equilibrium properties within GPU-minutes to hours per system -- closing the rare-event sampling gap that remained after the advent of diffusion-model equilibrium samplers.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
A simple explanation of “Enhanced Diffusion Sampling: Efficient Rare Event Sampling and Free Energy Calculation with Diffusion Models”
What is this paper about?
This paper is about making computer simulations of molecules—especially proteins—much faster and more reliable when the things we care about are rare and hard to see. The authors combine two ideas:
- Powerful AI models called diffusion models that can quickly make many realistic molecular shapes.
- Classic “enhanced sampling” tricks that temporarily nudge the system toward rare situations and then mathematically “undo” the nudge to get correct answers.
Together, this lets them study events like protein folding and compute free energies (a way to measure how stable something is) far more efficiently.
What questions are they trying to answer?
In simple terms, the paper asks:
- How can we quickly and accurately estimate important properties (like free energies) when the states that control those properties are very rare?
- Can we make diffusion models explore these rare states on purpose, yet still get unbiased, correct results for the real system?
- Can we turn classic enhanced sampling methods into tools that work with diffusion models and benefit from their speed?
How do they do it? (Methods in everyday language)
First, a bit of background:
- Molecular dynamics (MD) simulates atoms moving step by step, like watching a movie. It can get stuck for a long time in one “scene” (called slow mixing), and it almost never sees very rare scenes (rare states), so it can be slow.
- Diffusion models (like BioEmu) are AI generators. Instead of a movie, they “snap” independent pictures of likely molecular shapes. This avoids the slow-mixing problem, but rare states are still rarely generated.
The authors’ key idea is “bias and unbias,” done carefully:
- Bias (steering): Temporarily tilt the generator so it targets the rare regions you care about, much like pointing a camera at a hard-to-see spot. This is done at sampling time, without retraining the AI. Think of it like placing magnets that attract the sampler to certain features (for example, “more unfolded” proteins).
- Unbias (reweighting): After sampling, they mathematically correct (reweight) each sample so overall the results match the true, unbiased system. This is like oversampling rare neighborhoods in a survey and then dividing by a known factor so the city-wide statistics remain correct.
To make this concrete, they turn three classic enhanced sampling techniques into diffusion-model versions:
- UmbrellaDiff: Like setting up a series of soft “umbrellas” along a reaction coordinate (a simple measuring stick for progress, such as how folded a protein is). Each umbrella gently holds the sampler near a chosen region so the full range is covered. After sampling under all umbrellas, they combine results to get the true free-energy profile.
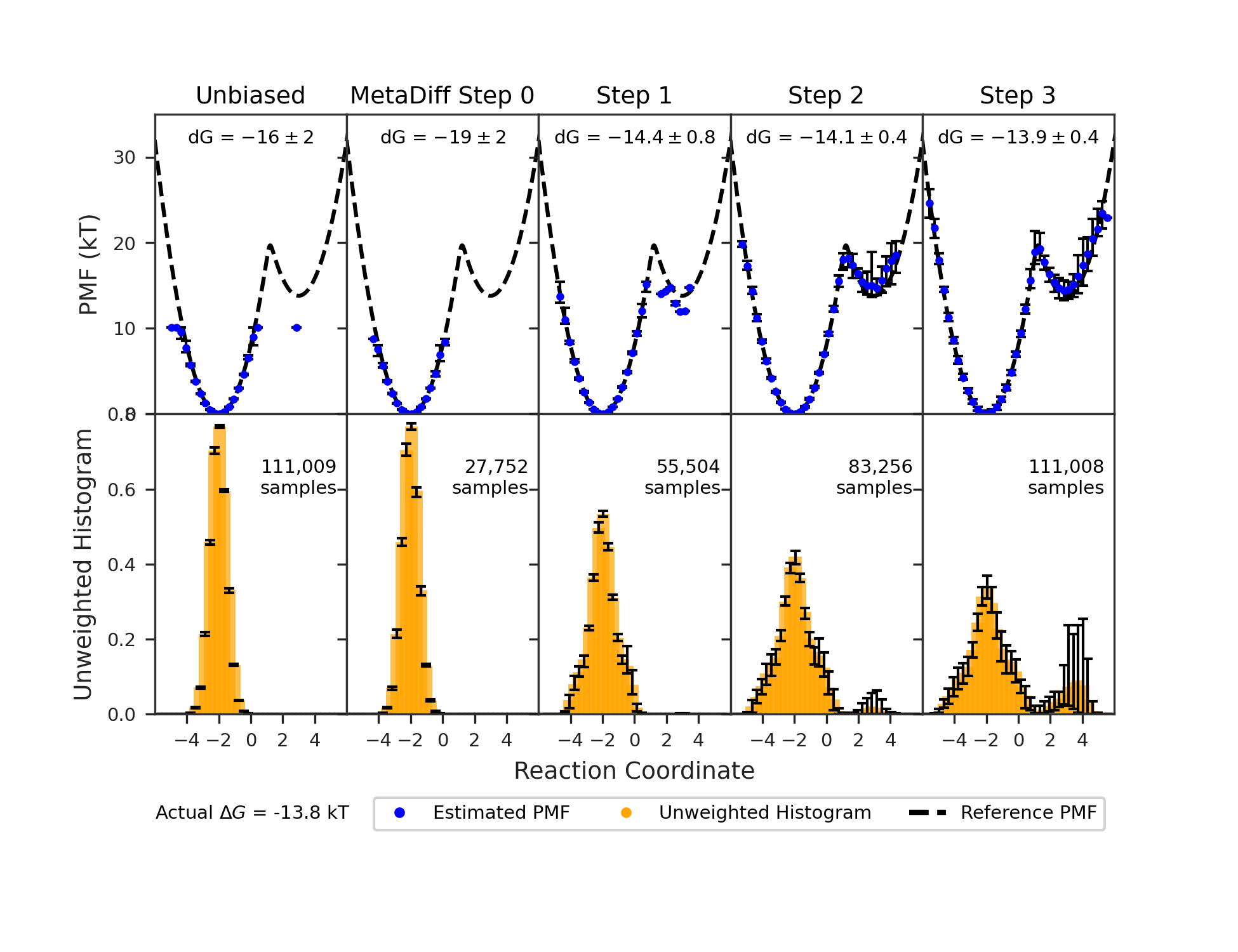
- MetaDiff: Like metadynamics, which places small “hills” in frequently visited areas to push exploration into new regions. Here, they add these hills batch-by-batch with the diffusion sampler, and they can combine everything on the fly to estimate the real free energy.
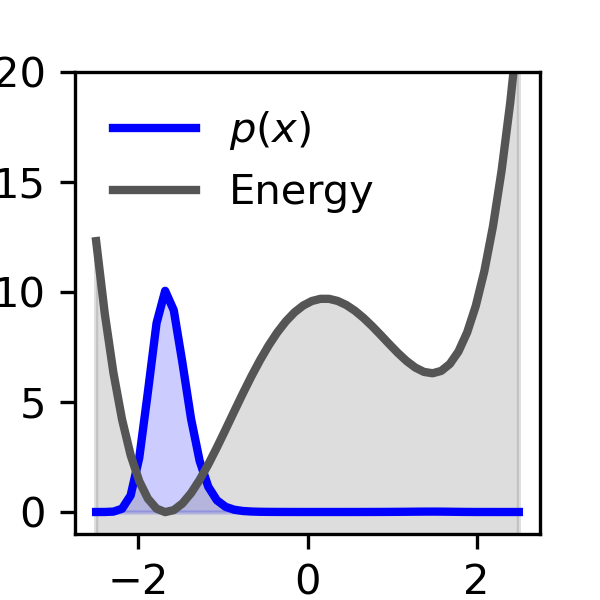
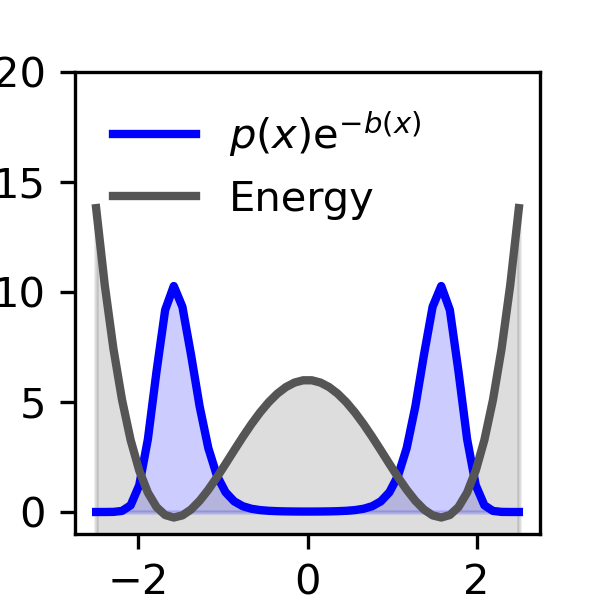
- ΔG-Diff: A method focused on differences in free energy between two states (for example, folded vs. unfolded). It “tilts” the sampling to focus on the important boundary between states and then computes the unbiased free-energy difference.
Behind the scenes, they use standard, well-trusted math tools (like MBAR/WHAM) to do the unbiasing and to combine data from multiple biased samplings in a way that minimizes error.
What did they find, and why is it important?
They test their approach on simple toy problems and on real protein systems. The main takeaways are:
- It’s fast: Instead of needing days or weeks, many calculations finish in minutes to hours on a single GPU.
- It’s accurate: Even when the states are very rare (for example, unfolded forms of a stable protein), the method recovers correct free energies.
- It solves both old problems at once: Diffusion models fix slow mixing, and the new “enhanced diffusion sampling” fixes rare-state estimation. Together, that closes a long-standing gap in molecular simulation.
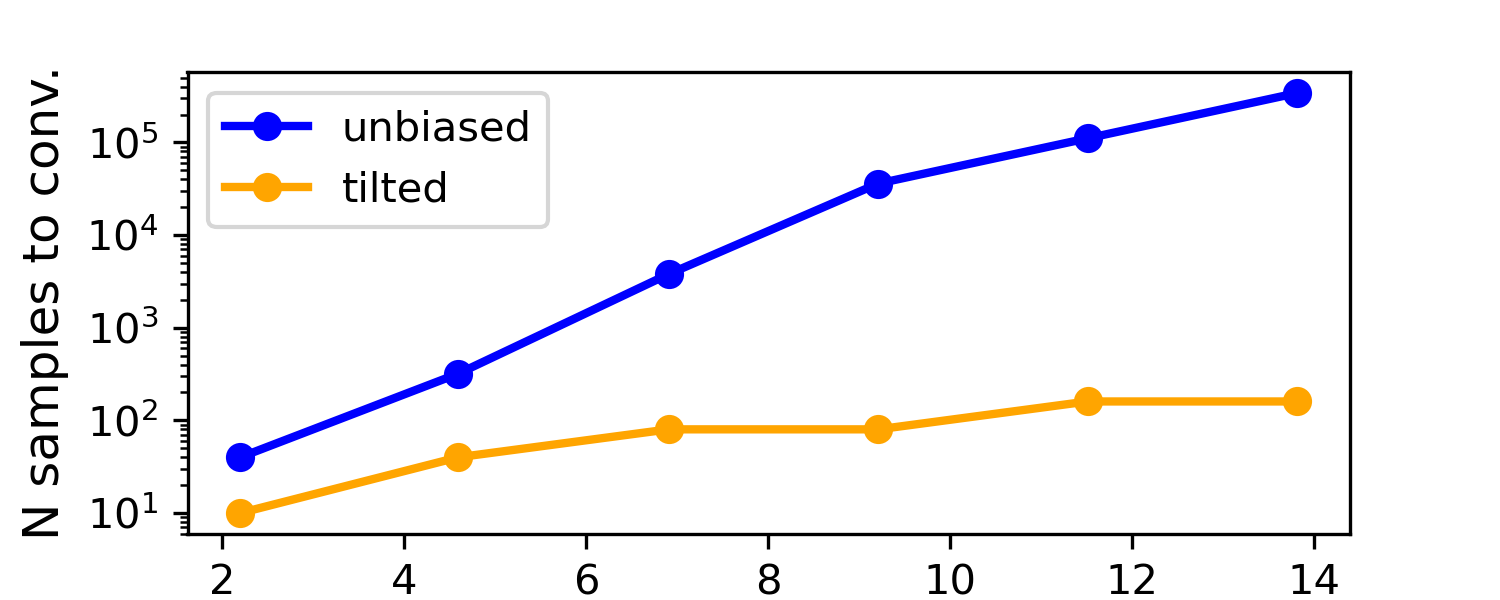
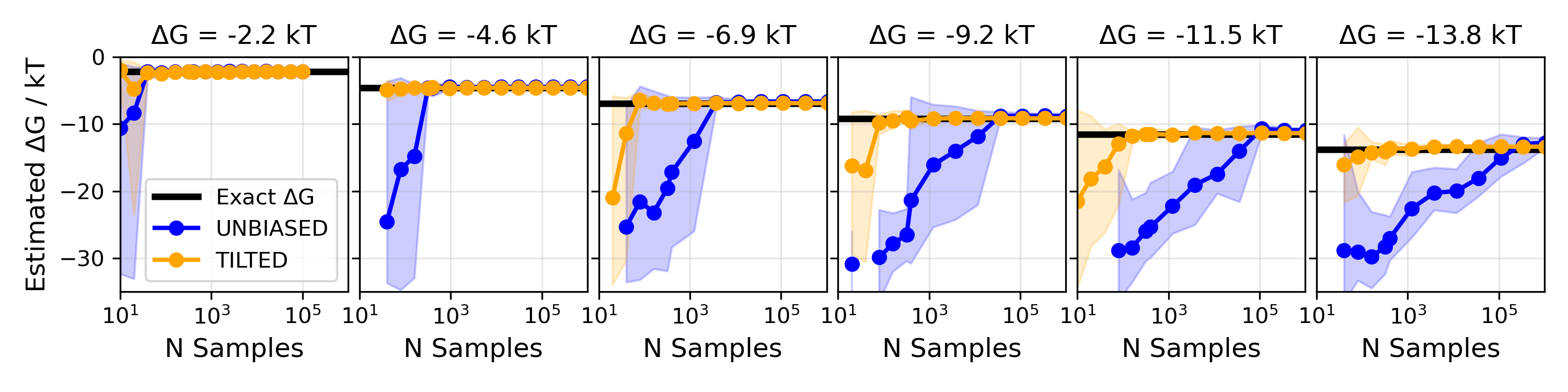
A simple example they show is a double-well landscape (imagine two valleys with different depths). With normal, unbiased sampling, you’d need a huge number of tries to see the rare valley enough times to measure its depth accurately. With their biased-and-unbiased method, the number of samples needed grows only slowly—even when the rare valley is extremely rare—making the job practical.
What could this change in the future?
This work could make tough molecular questions much more accessible:
- Protein folding and stability: Quickly estimating how stable a protein is, even when the unfolded state is extremely rare.
- Binding and conformational changes: Studying how drugs bind or how proteins switch shapes, which often involves rare events.
- Faster research cycles: Because it’s efficient and works with pretrained diffusion models, researchers can get reliable answers sooner, explore more ideas, and tackle larger systems.
In short, the paper shows a way to aim AI-driven molecular generators at rare, important events and still get unbiased, trustworthy results. This can accelerate discoveries in biology, chemistry, and drug design by turning previously impractical calculations into routine ones.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a consolidated list of concrete gaps, limitations, and unresolved questions that, if addressed, could strengthen and extend the framework and its applications.
- Assumption of a correct pretrained equilibrium distribution: The framework assumes the diffusion model’s output distribution p(x) matches the true equilibrium ensemble. There is no quantification of model bias, calibration of p(x) to physical Boltzmann weights, or correction procedures when the model misestimates energies or densities—especially critical for absolute free energies reported in kcal/mol.
- Impact of score/model errors on unbiasedness: Steering and reweighting rely on accurate reverse-time scores ∇ log p_t and the FKC weight dynamics. The paper does not analyze how score errors, discretization of the reverse SDE, or approximation of expectations (e.g., E_{x∼q_t}[F_t(x)]) propagate into estimator bias/variance, nor does it provide bounds or diagnostics to detect when unbiasedness is compromised.
- Weighted MBAR theory and practice: The weighted MBAR variant (using per-sample effective masses α_n derived from importance weights and per-window ESS) is proposed without a rigorous proof of asymptotic unbiasedness under correlated terminals or late branching, or guidance on optimal α_n choices. A formal treatment of conditions for correctness, variance inflation due to within-window correlations, and comparison to alternative weight normalizations is missing.
- Discretization and SMC stability: The Feynman–Kac steering uses discretized SDEs and resampling. There is no systematic study of time-step selection, resampling thresholds, particle impoverishment, and numerical stability under strong biases. Concrete guidance for avoiding weight degeneracy and ensuring high ESS across challenging bias schedules is needed.
- Estimation of E_{x∼q_t}[F_t(x)]: The weight evolution requires the expectation of F_t under q_t. The paper does not specify practical estimators (e.g., batch estimates, control variates) nor analyze variance, bias, and computational overhead of this term in high dimensions.
- Late branching and correlation handling: While late branching near t≈0 is recommended to increase sample count, there is no quantitative guidance on branching depth, induced correlations, how to adjust MBAR/uncertainty estimates, or when cluster/bootstrap resampling suffices. A principled procedure to balance branching gains versus correlation penalties remains open.
- Choice and discovery of collective variables (CVs): The methods still require user-chosen CVs (e.g., fraction of native contacts). There is no integrated strategy to learn or validate CVs within the diffusion framework (e.g., via VAMPnets, RAVE, DeepTICA), nor procedures to detect and mitigate hidden barriers in orthogonal coordinates when CVs are suboptimal.
- Multidimensional umbrella/metadynamics not demonstrated: Although the extension to multidimensional CVs is stated as straightforward, the paper only illustrates 1D cases. The scaling of steering, kernel choice (σ), overlap management, and MBAR conditioning in higher-dimensional CV spaces is untested and may suffer severe weight degeneracy.
- Parameter selection for MetaDiff: Concrete, generalizable guidelines for metadynamics kernel width σ, base-height schedule h_k, tempering factor γ, and batch size N_k are absent. Automated or adaptive tuning rules (e.g., using online MBAR overlap, EMUS error attribution, or cross-validation) are not provided.
- Online MBAR for history-dependent biases: MetaDiff uses evolving, history-dependent biases, but the paper does not detail an online MBAR pipeline (e.g., scheduling updates, window aggregation over time, weight reuse across iterations, and computational cost) nor assess how estimator variance behaves during early “fill-up” phases.
- Robustness to strong biases: The effect of overly strong umbrellas or metadynamics hills on FKC steering (e.g., loss of support, catastrophic weight collapse) is not characterized. Practical bounds on bias strength, adaptive tempering, or regularization strategies are needed.
- Differentiability of bias and CVs: Steering requires ∇b_t(x). For discrete or non-differentiable state indicators (e.g., folded/unfolded classifiers), the paper does not propose differentiable surrogates, smoothing strategies, or quantify the impact on steering accuracy.
- Out-of-distribution (OOD) rare states: If rare states are absent or underrepresented in training data, the diffusion model may not cover them; steering may fail to create truly novel, high-free-energy configurations. Methods to expand support (e.g., fine-tuning, hybrid energy-based models, rejection/repair steps) and to diagnose OOD sampling are not addressed.
- Physical validity of generated configurations: There is no systematic validation of steric, bonding, and chemical constraints under heavy biasing (e.g., avoidance of nonphysical structures), nor criteria to detect and discard invalid generations or repair them before reweighting.
- Applicability to explicit solvent systems: The framework’s performance and scalability in all-atom explicit solvent (with many degrees of freedom, slow solvent coupling, and high-dimensional CVs) is not demonstrated, despite being a key target. Memory, compute, and stability considerations for such systems remain unexplored.
- Computational scaling and cost model: Claims of “GPU-minutes to hours” lack detailed benchmarks versus MD (for comparable accuracy), scaling laws with system size, number of umbrellas/windows, dimensionality of CVs, and target free-energy differences. A cost–accuracy tradeoff analysis is missing.
- Uncertainty quantification under correlated, weighted samples: The use of MBAR covariance and bootstrap is suggested, but there is no comprehensive treatment for importance-weighted, resampled, and branched samples with within-window correlations. Guidance on reliable confidence intervals and error bars is needed.
- Integration with experimental constraints and energies: The framework does not show how to incorporate experimental restraints or physical energy evaluations (U(x)) during steering or reweighting, nor how to reconcile learned p(x) with energy-based models to anchor absolute thermodynamic scales.
- Automatic window placement and overlap control: UmbrellaDiff relies on manual or heuristic window centers and stiffness. There is no algorithm to automatically place/add/merge windows using overlap matrices, MBAR conditioning, or EMUS variance attribution, nor strategies to adaptively rebalance sampling effort.
- ΔG-Diff specification incomplete: The section outlining free-energy differences via tilted ensembles is truncated and lacks full algorithmic details (state definitions, path/bridging strategies, bias scheduling, reweighting, diagnostics), leaving the approach underspecified for practical use.
- Kinetics and rate estimation: The work focuses on equilibrium properties and free energies. It does not address whether (and how) diffusion models plus steering could yield kinetic observables (e.g., rates, transition pathways) or how to combine with TPT/MSM analysis under biased sampling.
- Robust folded/unfolded membership definitions: The χ(x) indicator function is mentioned but not standardized. There is no analysis of threshold sensitivity, fuzzy memberships, or validation against experimental folding markers to ensure reliable ΔG_fold estimation across diverse proteins.
- Reproducibility and implementation details: Key practical elements—time discretization schemes, resampling algorithms, control drift choices, score network architectures and training regimes, and code availability—are not specified, limiting reproducibility and independent validation.
- Combining with alchemical transformations: While classical FEP/TI are cited, the framework does not demonstrate or detail how enhanced diffusion sampling would handle alchemical free energies (e.g., protein–ligand RBFE) with large conformational rearrangements, nor propose hybrid schemes (AIS/SMC bridges, staged alchemy with steering).
- Diagnostics for model–data consistency: Beyond MBAR overlap and ESS, there are no diagnostics to detect when steering/reweighting estimates disagree with independent MD or experimental baselines due to model misspecification, nor corrective workflows (e.g., reweighting the generative model itself, retraining, or incorporating physics-based constraints).
Practical Applications
Overview
This paper introduces Enhanced Diffusion Sampling—steering pretrained diffusion-model equilibrium samplers to efficiently sample rare-event regions while preserving unbiased thermodynamic estimators via exact reweighting. It instantiates the framework in three deployable algorithms: UmbrellaDiff (umbrella sampling with diffusion models), MetaDiff (a batched analogue of metadynamics), and ΔG‑Diff (free-energy differences via tilted ensembles). The methods leverage Feynman–Kac Corrector (FKC) steering, effective sample-size (ESS) diagnostics, resampling, and weighted MBAR unbiasing to deliver fast, accurate free-energy landscapes and observables in GPU minutes to hours per system.
Below are the practical, real-world applications derived from the paper’s findings, methods, and innovations. The items are grouped into immediate and long-term applications, with sector linkages, candidate tools/workflows, and explicit assumptions/dependencies.
Immediate Applications
These applications can be deployed now, contingent on having suitable pretrained diffusion models (e.g., BioEmu) and implementing the steering/unbiasing workflows described.
- Rapid estimation of protein folding free energies (ΔG_fold) for construct prioritization
- Sectors: healthcare/biotech; academia
- Tools/workflows: ΔG‑Diff or UmbrellaDiff with membership functions (e.g., fraction of native contacts) and weighted MBAR; GPU pipeline to get ΔG_fold within minutes–hours; integrated ESS and overlap diagnostics to ensure reliability
- Uses: thermostabilization, protein engineering (identify stabilizing mutations), biotherapeutics developability screening
- Assumptions/dependencies: availability and fidelity of a pretrained diffusion model covering the protein’s equilibrium ensemble; differentiable order parameters; sufficient window overlap; unbiasedness relies on correct FKC steering and MBAR weight computation
- Fast PMF computation along chosen reaction coordinates (e.g., end-to-end distance, fraction of native contacts)
- Sectors: healthcare/biotech; academia
- Tools/workflows: UmbrellaDiff with harmonic umbrellas and MBAR/WHAM; automated window placement with overlap checks; kernel-based density estimates; late branching near t≈0 to increase sample count
- Uses: mapping folding pathways, conformational transitions, loop motions, domain rearrangements
- Assumptions/dependencies: meaningful collective variable (CV) that captures relevant progress; sufficient coverage of low-probability regions via umbrellas; stable MBAR convergence
- Batchwise metadynamics for exploration of undersampled states with online unbiased estimators
- Sectors: healthcare/biotech; academia
- Tools/workflows: MetaDiff with well-tempered bias accumulation (Gaussian kernels), batched updates, and online MBAR to report unbiased PMFs at each iteration; stopping criteria via bias/PMF change thresholds
- Uses: discovering alternative pathways/minima, rapidly “filling” free-energy wells while retaining unbiased estimates at each stage
- Assumptions/dependencies: appropriate kernel width and tempering factor; robust steering to each bias condition; reliable ESS/resampling to control variance
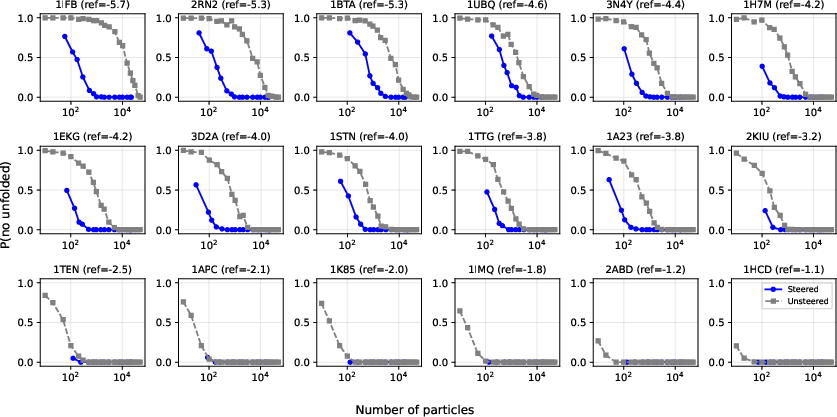
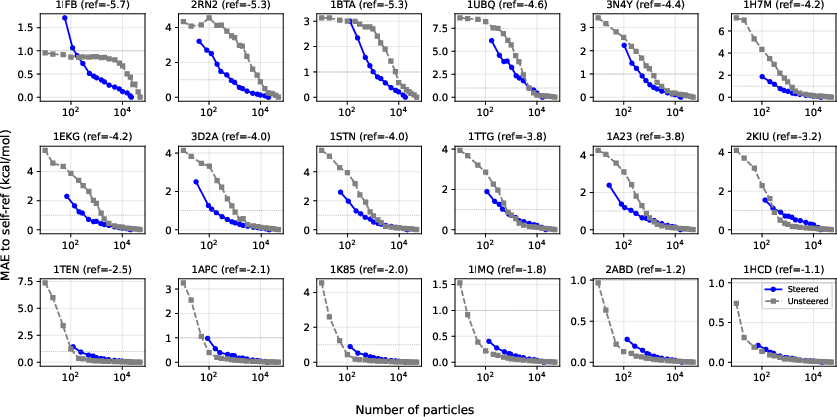
- Overcoming kinetic trapping in umbrella sampling via iid steered diffusion generations
- Sectors: healthcare/biotech; academia
- Tools/workflows: UmbrellaDiff steering to draw iid samples per umbrella; MBAR overlap matrix diagnostics to confirm connectedness; EMUS variance analysis to reallocate sampling
- Uses: compute PMFs even when orthogonal slow modes create hidden barriers; bypass hysteresis seen in MD-based umbrellas
- Assumptions/dependencies: unbiased diffusion model sampling (iid terminals); adequate umbrella design and overlap; accurate reweighting with weighted MBAR
- Efficient conformational landscape mapping for medium-sized proteins (≈50–200 residues)
- Sectors: healthcare/biotech; academia
- Tools/workflows: UmbrellaDiff/MetaDiff pipelines with BioEmu-like samplers; path coverage using CVs with adaptive umbrella stiffness; ESS-driven adaptive sampling
- Uses: allosteric regulation analysis, cryptic pocket discovery, conformational selection vs. induced fit hypotheses
- Assumptions/dependencies: pretrained model capturing relevant conformational heterogeneity; suitable CVs; stable computational performance for target size and solvent model used by the emulator
- Experimental ensemble refinement and consistency checks
- Sectors: academia; structural biology
- Tools/workflows: steering to match experimental constraints (e.g., CVs informed by NMR/smFRET), apply MetaDiff to broaden coverage, and MBAR to reweight to equilibrium; compare PMFs/ΔG with experimental measurements
- Uses: hypothesis testing, reconciling cryo-EM states/populations with thermodynamic predictions
- Assumptions/dependencies: availability of suitable experimental observables; emulator trained/fine-tuned on relevant data; careful mapping of observables to CVs and membership functions
- Integration into existing free-energy analysis stacks
- Sectors: software; academia; industry R&D
- Tools/workflows: Python libraries wrapping FKC steering, ESS/resampling, weighted MBAR; adapters to PyMBAR, visualization utilities for overlap matrices, automated window placement and diagnostics
- Uses: plug-and-play enhanced sampling with diffusion models in research pipelines; reproducible thermodynamic estimation
- Assumptions/dependencies: correct implementation of weighted MBAR; robust numerical solvers for fixed-point equations; correlation-aware uncertainty quantification (cluster bootstrap)
- Compute and sustainability benefits in simulation-heavy organizations
- Sectors: policy (R&D resource planning); industry R&D; academia HPC
- Tools/workflows: replacing long MD trajectories with GPU-minutes to hours diffusion-steering runs; track energy savings and throughput improvements
- Uses: lower carbon footprint and cost per thermodynamic estimate; expanded coverage of targets and variants
- Assumptions/dependencies: validated accuracy on internal benchmarks; existing GPUs compatible with diffusion inference; governance around ML-based thermodynamics
- Educational and training modules for enhanced sampling
- Sectors: education; academia
- Tools/workflows: teaching labs demonstrating iid sampling vs. rare-event steering; assignments comparing UmbrellaDiff/MetaDiff to MD-based umbrellas/metadynamics
- Uses: modern pedagogy in computational biophysics and statistical mechanics; reproducible exercises
- Assumptions/dependencies: access to pretrained models and tutorial datasets; simplified CVs for classroom use
Long-Term Applications
These applications require further research, scaling, validation, broader model availability, or extended engineering to be practical.
- Scalable binding free-energy predictions robust to large conformational changes
- Sectors: healthcare/drug discovery; biotech
- Tools/workflows: ΔG‑Diff across binding progress coordinates; MetaDiff to discover binding pathways; UmbrellaDiff to ensure overlap across intermediate states; reweighting without explicit evaluation of u(x)
- Potential products: cloud service for binding ΔG across diverse targets; automated conformational pathway discovery modules
- Assumptions/dependencies: pretrained emulators that jointly model protein–ligand and solvent ensembles with sufficient fidelity; validated CVs for binding progress; ground-truth benchmarks; handling chemical changes (alchemical RBFE) may need hybridization with energy-based models
- Generalized rare-event sampling in materials science (defects, phase transitions, nucleation)
- Sectors: materials; energy; catalysis
- Tools/workflows: UmbrellaDiff/MetaDiff on generative emulators of materials configurations; PMFs for transition pathways (e.g., ion migration, defect formation)
- Potential products: materials discovery pipelines that rapidly compute free-energy barriers and phase stabilities
- Assumptions/dependencies: diffusion-model emulators trained on relevant materials MD/DFT datasets; CVs for progress coordinates in solids; validation against experiments or high-level simulations
- Reaction free energies and transition-path sampling in complex environments
- Sectors: catalysis; energy storage; chemical engineering
- Tools/workflows: ΔG‑Diff with reaction progress CVs; MetaDiff to discover alternative mechanisms; umbrella windows for solvent/active-site coordinates
- Potential products: workflow components for catalytic cycle optimization and electrolyte reaction screening
- Assumptions/dependencies: high-fidelity emulators with explicit solvent/electrode environments; CVs capturing reaction coordinate and key solvent/ion couplings; benchmarking vs. enhanced MD and experiments
- Automated CV discovery and bias design via adaptive/learning-based strategies
- Sectors: software; academia; industry R&D
- Tools/workflows: coupling to RAVE/DeepTICA/VAMPnets-like methods for CV learning, then apply UmbrellaDiff/MetaDiff with adaptive window placement and EMUS variance-driven sampling allocations
- Potential products: “push-button” enhanced diffusion sampling platform with automatic CV selection and diagnostics
- Assumptions/dependencies: robust CV learning for explicit-solvent systems; stable interaction between learned CVs and steering; failure-mode detection when CVs are inadequate
- Large-scale coverage of membrane proteins, GPCRs, multimeric assemblies
- Sectors: healthcare/drug discovery; structural biology
- Tools/workflows: enhanced diffusion sampling on emulators that include membranes and multiple chains; multi-CV umbrellas for activation, oligomerization, or gating; online MBAR across many bias states
- Potential products: target-wide conformational atlas; activation energy profiling; ligand bias quantification
- Assumptions/dependencies: emulators trained to capture membrane/protein interactions; CVs for activation/gating; compute scaling and memory management for large systems
- Integration with energy-based diffusion models to tighten statistical efficiency
- Sectors: software; academia
- Tools/workflows: using models where u(x) is evaluable to refine steering and reweighting; hybrid AIS/SMC–MALA correctors; variance reduction techniques
- Potential products: general-purpose thermodynamics engine that unifies unbiased sampling with enhanced steering and exact energy evaluation
- Assumptions/dependencies: reliable energy-based models for complex biomolecules; careful algorithmic tuning; validation of unbiased estimators and uncertainty quantification
- Standardization and regulatory pathways for ML-driven thermodynamics
- Sectors: policy/regulatory; healthcare
- Tools/workflows: community benchmarks, reporting standards (ESS, overlap matrices, uncertainty), validation against experimental ΔG and PMFs; guidance documents for model risk management
- Potential products: best-practice protocols for submission-quality computational evidence in drug development
- Assumptions/dependencies: cross-stakeholder adoption; transparent datasets and methods; robust calibration/uncertainty reporting
- Cross-domain rare-event inference with generative models (beyond molecular systems)
- Sectors: risk analysis; finance; reliability engineering
- Tools/workflows: adapting FKC steering and MBAR-like reweighting to sample tails of complex generative posteriors; computing probability ratios across states
- Potential products: tail-risk simulators for complex systems; reliability estimators for engineered systems
- Assumptions/dependencies: existence of accurate generative models for target domains; mapping of domain-specific “states” and CVs; theoretical guarantees and practical diagnostics analogous to ESS/overlap
- Human-in-the-loop platforms for experiment design and prioritization
- Sectors: healthcare/biotech; materials
- Tools/workflows: interactive dashboards combining PMFs/ΔG with confidence intervals; suggest next mutations/constructs/windows to sample based on EMUS/MBAR variance contributions
- Potential products: decision-support tools for R&D teams to reduce experimental cycles
- Assumptions/dependencies: reliable uncertainty quantification; intuitive CV definitions for users; integration with LIMS and experiment tracking
General Assumptions and Dependencies (common across items)
- Pretrained diffusion-model emulators that faithfully approximate the equilibrium ensemble p(x) for the system of interest; coverage of relevant rare states is essential.
- Differentiable and physically meaningful CVs/progress coordinates; appropriate membership functions for state classification (e.g., folded/unfolded).
- Correct implementation of FKC steering, ESS monitoring/resampling, and weighted MBAR unbiasing; sufficient window overlap and connectedness.
- Robust uncertainty quantification (MBAR covariance or cluster bootstrap) and diagnostics (overlap matrices, EMUS variance, stability under increased sampling).
- Compute resources compatible with diffusion inference (GPUs); scaling considerations for larger systems or more complex environments.
- External validation against MD and experiment for critical applications (drug development, materials deployment) and governance around ML model accuracy and domain shift.
Glossary
- Annealed importance sampling (AIS): A method for estimating ratios of partition functions by gradually transforming one distribution into another without resampling. "annealed importance sampling (AIS) appears as the no-resampling special case"
- Boltzmann emulators: Generative models trained on MD data (and optionally experimental observables) to approximate equilibrium distributions and produce independent samples. "Boltzmann emulators approximate the equilibrium distribution by training on MD simulation data and fine-tuning on experimental observables."
- Boltzmann Generators: Flow-based generative models that sample from Boltzmann distributions defined by energy functions, aiming for independent equilibrium configurations. "Boltzmann Generators are flow-based approaches that aim to generate independent samples from a Boltzmann distribution defined via an energy function"
- Collective variables (CVs): Low-dimensional functions of configuration used to bias or analyze sampling, often capturing progress along a process. "periodically deposit repulsive hills (typically Gaussians) in the space of chosen collective variables (CVs) "
- Effective sample size (ESS): A measure of the efficiency of weighted samples, reflecting the number of equivalent independent samples. "We track the statistical efficiency of a weighted batch of samples using the Kish effective sample size (ESS)"
- EMUS: An error analysis framework for umbrella sampling that identifies windows contributing most to variance to guide adaptive sampling. "we optionally complement MBAR with EMUS error analysis to identifying windows that dominate variance and guide adaptive allocation of sampling effort"
- Energy-based diffusion models: Diffusion models where the underlying probability density and dimensionless energy can be efficiently evaluated. "Energy-based diffusion models, for which and can be efficiently evaluated, have been explored recently"
- Feynman–Kac (FK) potentials: Path-integral weighting potentials used in SMC/AIS to account for bias along reverse trajectories. "via Sequential Monte Carlo (SMC) with Feynman–Kac (FK) potentials—keeping the proposal dynamics unchanged and accounting for the bias with incremental importance weights along the reverse trajectory"
- Feynman–Kac Corrector (FKC): A steering methodology that modifies diffusion reverse dynamics and accumulates weights to target biased marginals. "Subsequently, we employ the Feynman-Kac Corrector (FKC) biased sampling methodology"
- Free energy perturbation (FEP): A technique to compute free energy differences by reweighting samples from one thermodynamic state to another. "free energy perturbation (FEP) \cite{Zwanzig_JCP54_TI}"
- Importance weights: Factors attached to samples to correct for discrepancies between proposal and target distributions. "The weights are importance weights, encoding the discrepancy between proposal and target biased marginals."
- Kernel density estimator (KDE): A nonparametric method for estimating probability densities from samples via smooth kernels. "Practically, we obtain PMFs by constructing a weighted histogram or kernel density estimator from the weighted sample"
- Markov state model (MSM): A framework that models long-timescale dynamics via metastable states and transition probabilities, revealing slow relaxation spectra. "these systems typically exhibit a spectrum of slow relaxation processes rather than a single dominant timescale, as explored in the MSM literature"
- Metadynamics: An enhanced sampling method that adds history-dependent repulsive hills in CV space to escape free-energy minima and estimate free energies. "Metadynamics \cite{LaioParrinello_PNAS99_12562,LaioGervasio_RPP08_Metadynamics,BarducciBonomiParrinello_WIREs11} is a widely used enhanced sampling method"
- Metropolis–adjusted Langevin algorithm (MALA): A Markov chain Monte Carlo corrector that mixes samples within a target marginal by proposing Langevin steps with Metropolis acceptance. "Invariant correctors (e.g., Metropolis–adjusted Langevin, MALA) that are interleaved with the reverse dynamics to mix within the current biased marginal"
- Multistate Bennett acceptance ratio (MBAR): A statistically optimal estimator that combines samples from multiple biased states to produce minimum-variance, unbiased expectations. "multistate Bennett acceptance ratio (MBAR) method"
- Partition function: The normalization constant of a probability distribution over configurations, crucial for defining biased ensembles. "Z is an unknown normalization constant (partition function) that we will not need to evaluate explicitly."
- Potential of mean force (PMF): The free-energy profile along a reaction coordinate, derived from the marginal probability density. "free energy profile—also known as potential of mean force (PMF)"
- Reaction coordinate: A function that tracks progress along a molecular process, used to bias sampling and compute PMFs. "along a user-chosen reaction coordinate "
- Replica exchange molecular dynamics (REMD): An enhanced sampling approach that swaps configurations between simulations at different temperatures to accelerate mixing. "hybrids such as REMD+metadynamics"
- Score guidance: A technique that modifies the reverse-time score by adding gradients (e.g., from classifiers or rewards) to steer generations. "Score guidance, which alters the reverse-time score by adding the gradient of a classifier, constraint, or reward to tilt generations toward desired properties"
- Sequential Monte Carlo (SMC): A family of particle methods that propagate and resample weighted ensembles to target evolving distributions. "via Sequential Monte Carlo (SMC) with Feynman–Kac (FK) potentials"
- Stochastic differential equation (SDE): A differential equation with stochastic terms (noise) governing the forward corruption and reverse denoising dynamics in diffusion models. "We assume our diffusion model has been trained to reverse the corruption process defined by a stochastic differential equation"
- Stratified sampling: A resampling scheme that reduces variance by dividing the weight distribution into strata and sampling proportionally. "During denoising we periodically resample the particles using stratified sampling."
- Umbrella sampling: An enhanced sampling technique that applies restraining bias potentials across windows to ensure coverage and overlap along a coordinate, then reweights to recover equilibrium properties. "Here we describe how the classical umbrella sampling method \cite{Torrie_JCompPhys23_187} can be adapted to steered diffusion models."
- Weighted histogram analysis method (WHAM): A technique that combines biased histograms to estimate unbiased densities and free energies. "unbiasing methods such as WHAM"
- Wiener process: A mathematical model of continuous-time Gaussian noise used in SDEs for diffusion models. "Here is a standard Wiener process."
- Well-tempered metadynamics: A metadynamics variant where hill heights are tempered so the bias converges to a scaled negative free energy. "in well-tempered metadynamics the hill heights are tempered so that the bias converges (up to a known factor) to the negative free energy"
Collections
Sign up for free to add this paper to one or more collections.