PackFlow: Generative Molecular Crystal Structure Prediction via Reinforcement Learning Alignment
Abstract: Organic molecular crystals underpin technologies ranging from pharmaceuticals to organic electronics, yet predicting solid-state packing of molecules remains challenging because candidate generation is combinatorial and stability is only resolved after costly energy evaluations. Here we introduce PackFlow, a flow matching framework for molecular crystal structure prediction (CSP) that generates heavy-atom crystal proposals by jointly sampling Cartesian coordinates and unit-cell lattice parameters given a molecular graph. This lattice-aware generation interfaces directly with downstream relaxation and lattice-energy ranking, positioning PackFlow as a scalable proposal engine within standard CSP pipelines. To explicitly steer generation toward physically favourable regions, we propose physics alignment, a reinforcement learning post-training stage that uses machine-learned interatomic potential energies and forces as stability proxies. Physics alignment improves physical validity without altering inference-time sampling. We validate PackFlow's performance against heuristic baselines through two distinct evaluations. First, on a broad unseen set of molecular systems, we demonstrate superior candidate generation capability, with proposals exhibiting greater structural similarity to experimental polymorphs. Second, we assess the full end-to-end workflow on two unseen CSP blind-test case studies, including relaxation and lattice-energy analysis. In both settings, PackFlow outperforms heuristics-based methods by concentrating probability mass in low-energy basins, yielding candidates that relax into lower-energy minima and offering a practical route to amortize the relax-and-rank bottleneck.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
Overview
This paper introduces PackFlow, a smart computer system that helps scientists predict how molecules arrange themselves in a solid crystal. This matters for things like making better medicines or flexible phone screens, because the way molecules pack together can change how a material behaves (for example, how it dissolves, how stable it is, or how well it carries electricity).
Key Objectives
The paper focuses on three simple goals:
- Create a tool that can quickly suggest realistic crystal structures for a given molecule.
- Make those suggestions physically sensible so they are more likely to be stable.
- Reduce the time and computer power needed later to check and refine these suggestions.
Methods and Approach
Think of building a crystal like packing identical toy blocks (molecules) into a repeating 3D pattern (the crystal). Two big decisions define that pattern:
- Where each atom sits (its 3D position).
- The shape and size of the “room” they repeat in (the crystal’s unit cell, also called the lattice).
Here’s how PackFlow works, explained in everyday terms:
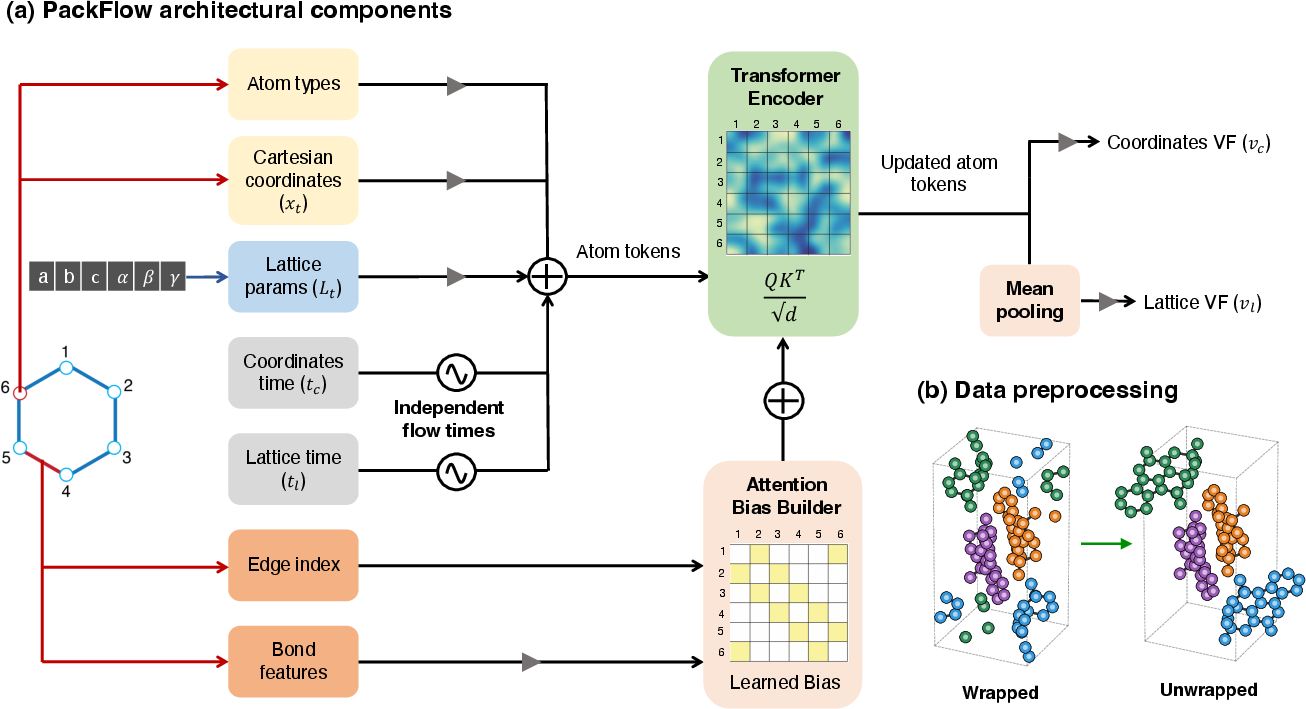
- Joint generation of positions and the “room”: PackFlow predicts both the atom positions and the unit cell at the same time. That’s like arranging furniture and deciding the room’s shape together, so everything fits better.
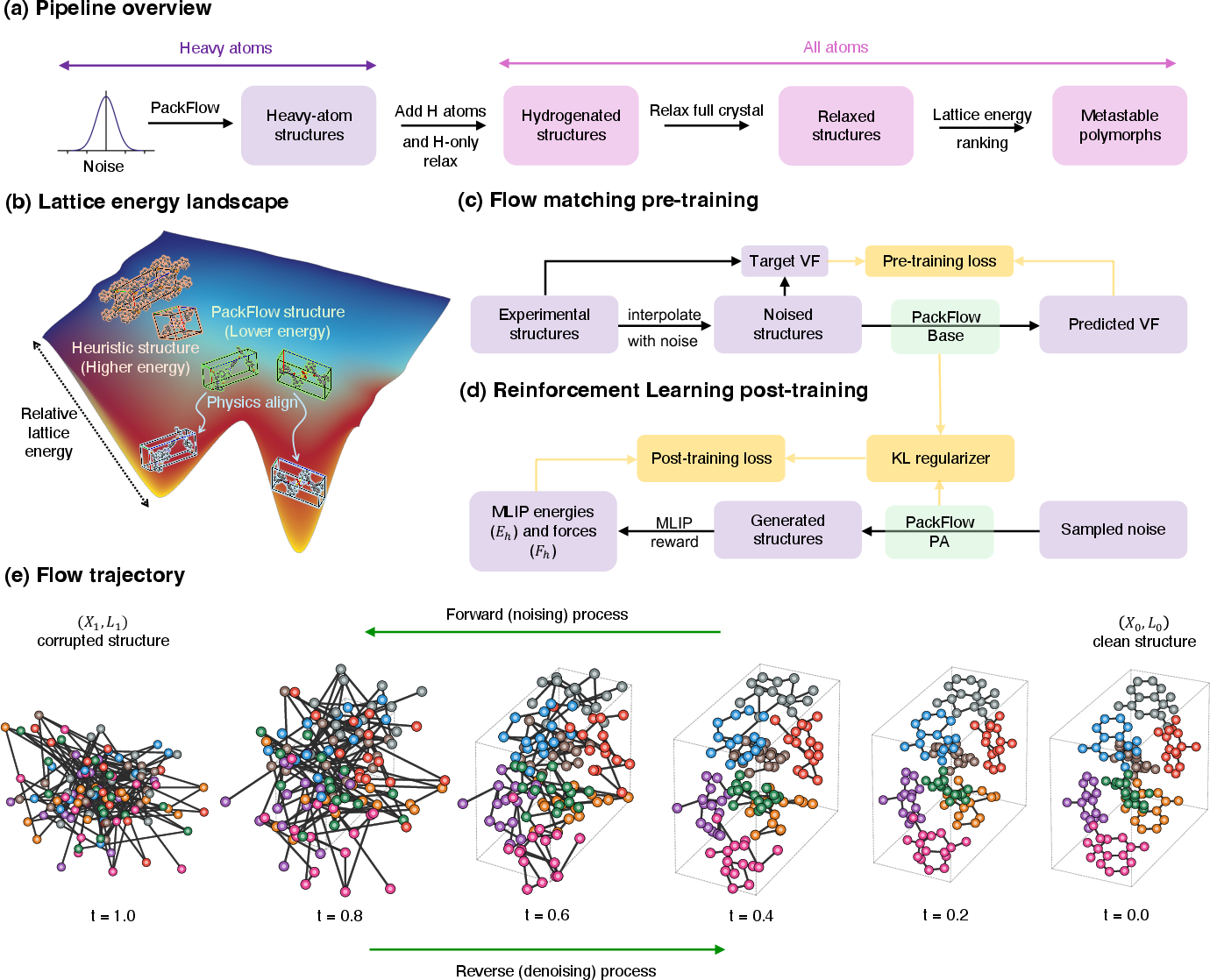
- Heavy atoms first: It starts by placing the heavier atoms (like carbon, nitrogen, oxygen). Lighter hydrogen atoms are added afterward and adjusted quickly, because heavy atoms mostly decide the overall structure.
- Flow matching (turning noise into structure): PackFlow learns to transform random noise into realistic crystals step by step, like sculpting a statue by gradually removing clay until a shape appears. This process is called “flow matching,” and it’s guided by examples of real crystals.
- Bond-aware attention (don’t break the molecule): The model keeps track of which atoms are bonded inside the molecule, so it doesn’t put bonded atoms impossibly close or far. It uses a transformer (a type of neural network) that pays extra attention to bonded atom pairs—similar to prioritizing close friends in a group conversation.
- Independent timing for atoms and lattice (different rhythms): The model uses separate “clock speeds” for cleaning up atom positions and the unit cell shape, because they benefit from different paces. It’s like tuning the furniture layout and room dimensions on different schedules.
- Unwrapping at the edges (no awkward cuts): Crystals repeat in all directions, so molecules can appear split across boundaries. PackFlow “unwraps” molecules to keep them intact while learning, avoiding weird chopped bonds.
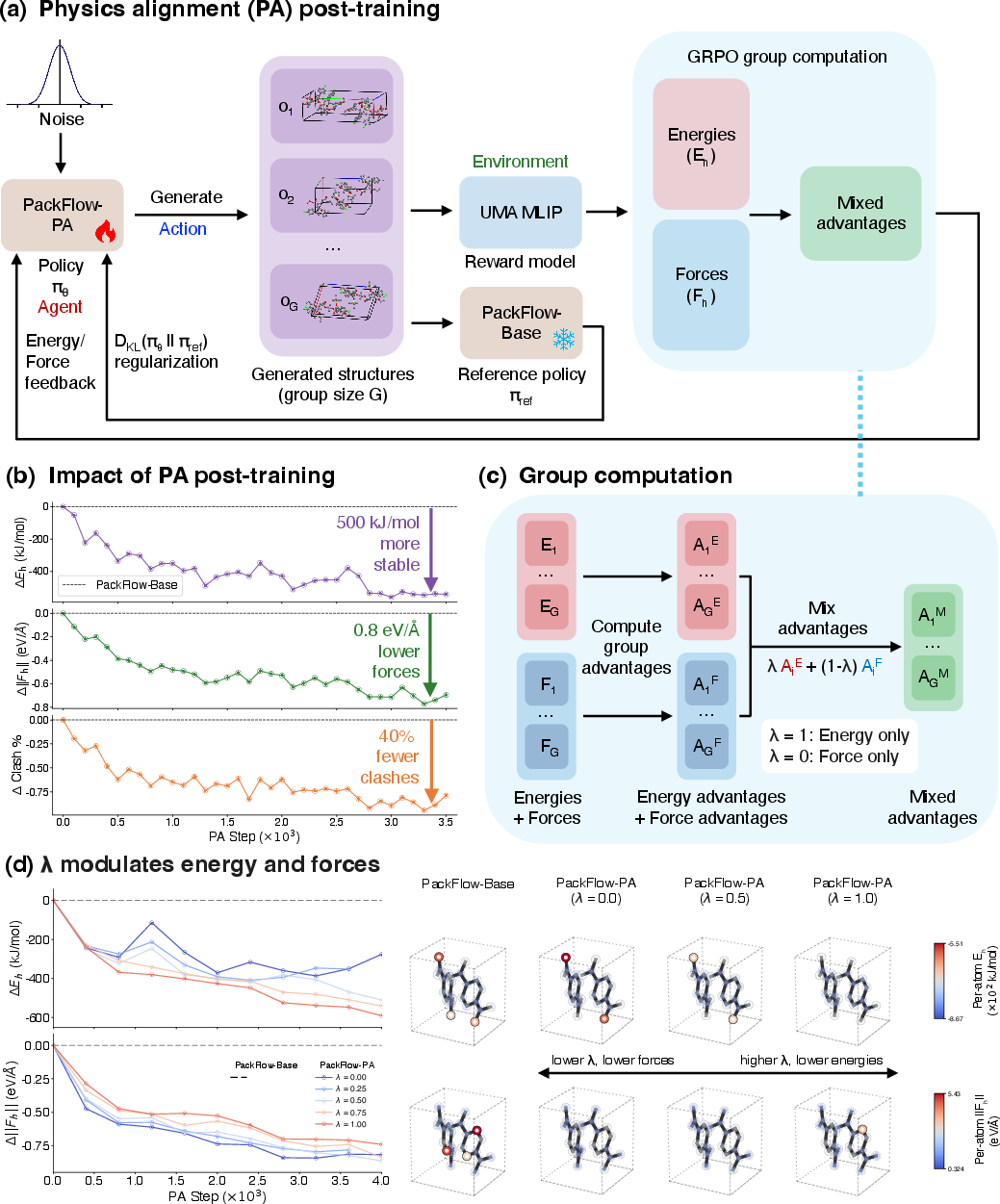
- Physics alignment via reinforcement learning (practice with feedback): After pre-training, PackFlow practices generating crystals and gets feedback from a fast physics calculator (a machine-learned interatomic potential, or MLIP) that estimates:
- Energy: Lower energy usually means more stable.
- Forces: Lower forces mean the structure isn’t under strain.
- The model uses reinforcement learning (like a game where it gets points for good moves) to adjust its suggestions toward lower energy and lower force configurations. This is called “physics alignment.”
- GRPO and advantage mixing (fair comparisons within each case): The RL method compares multiple generated options for the same molecule and favors the better ones. It mixes energy and force feedback in a balanced way by normalizing them first, so one signal doesn’t overwhelm the other. This reduces fiddly tuning.
Main Findings
The authors tested PackFlow against a well-known method called Genarris (which uses smart random generation and filtering). Here’s what they found:
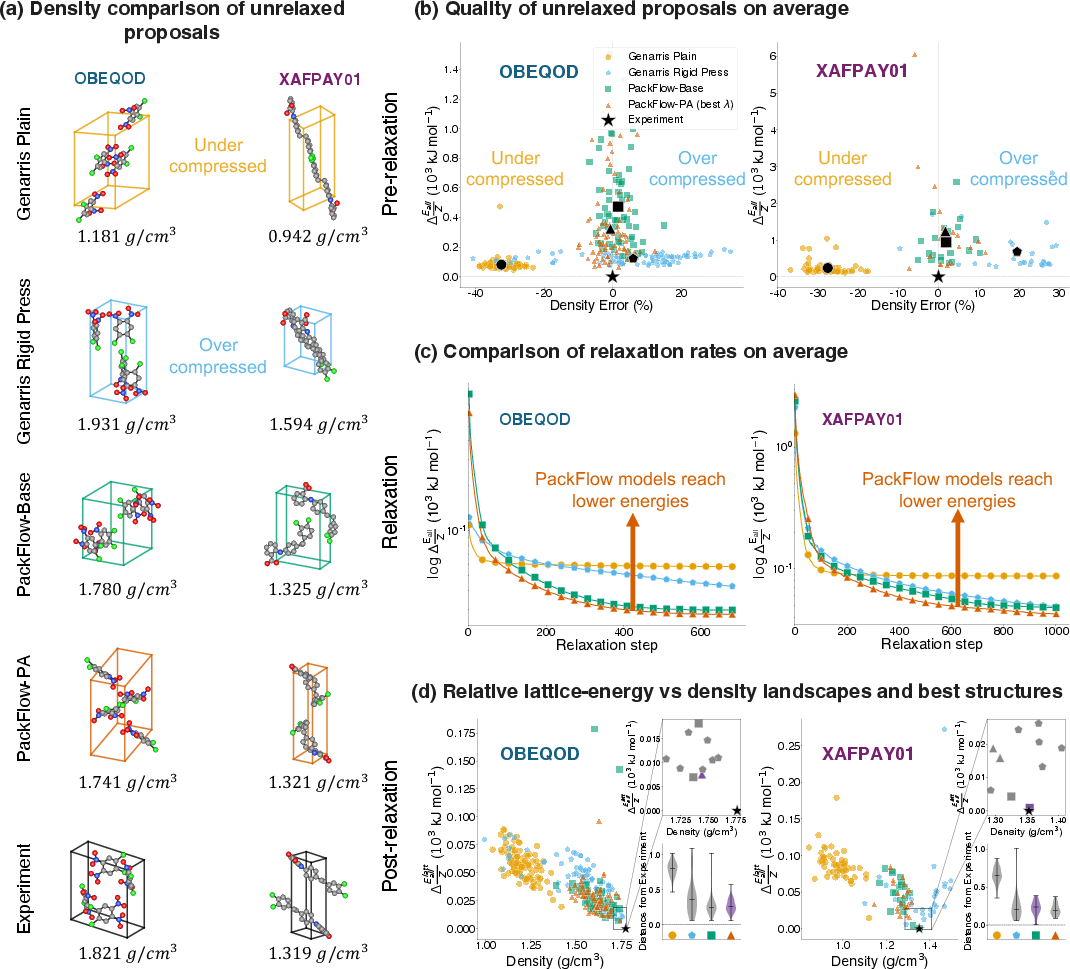
- Better initial guesses:
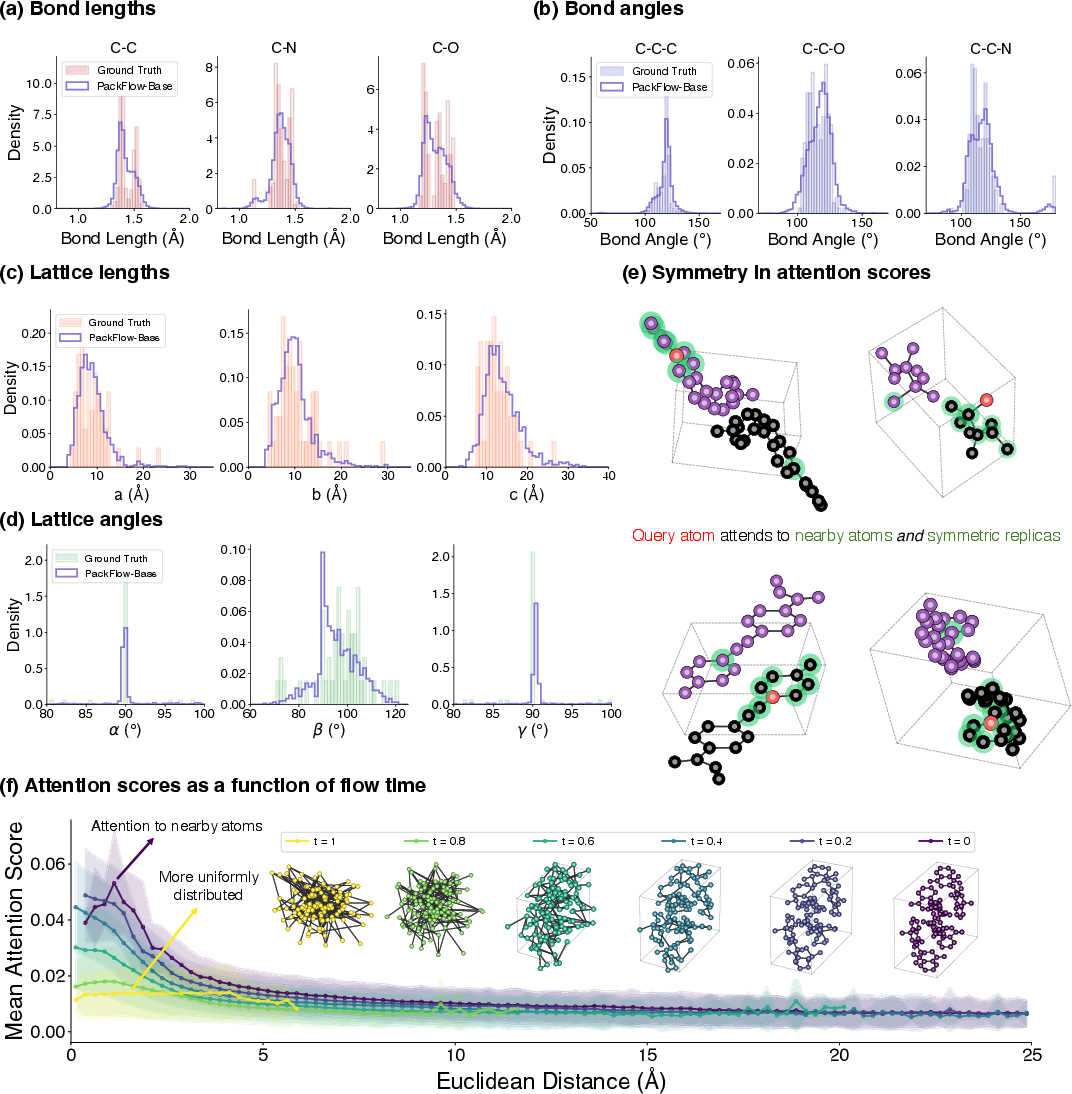
- PackFlow’s suggested structures have densities (how tightly atoms pack) much closer to real experimental crystals.
- Fewer “clashes” where atoms overlap too much (a clear sign of something physically wrong).
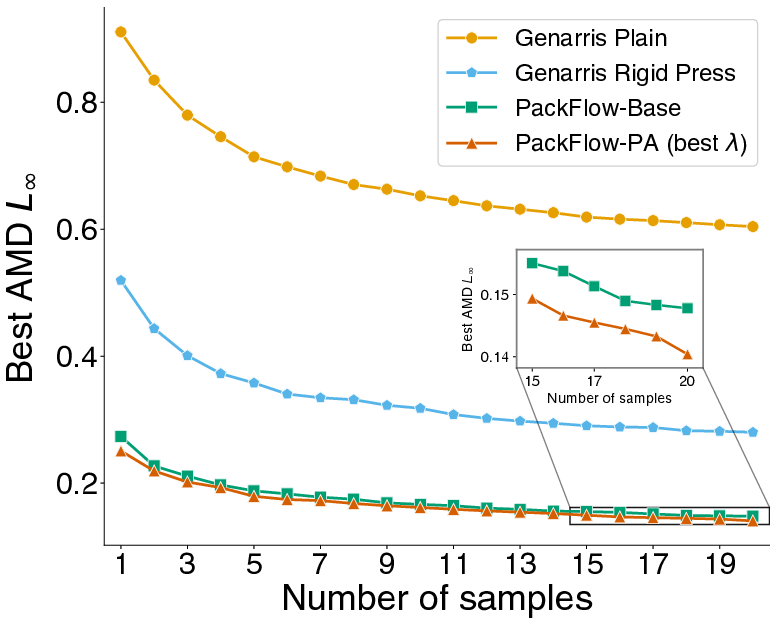
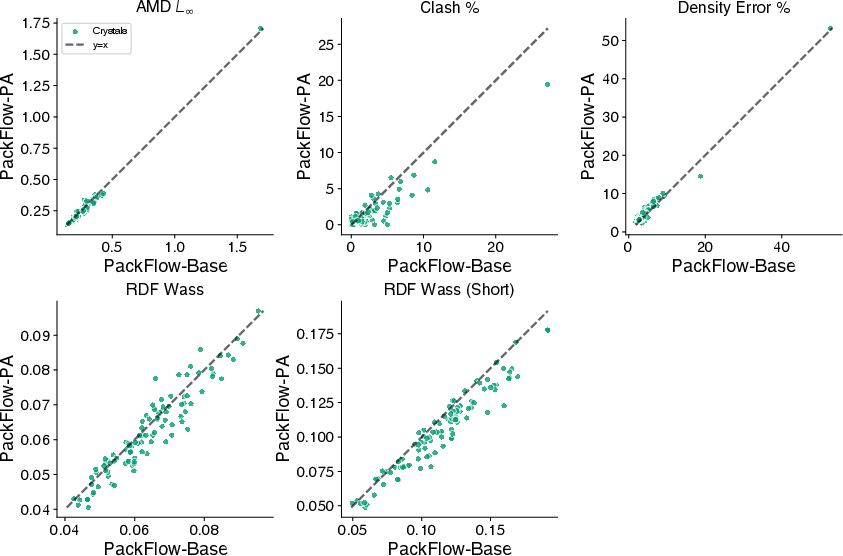
- Higher similarity to known structures, measured using simple shape-and-distance comparisons (AMD and RDF metrics).
- Post-training improvements:
- Physics alignment further reduces clashes and nudges structures into lower-energy regions without slowing down generation.
- Faster generation:
- PackFlow can produce one candidate in roughly a tenth of a second on a single GPU during their tests, which is fast.
- End-to-end wins in real challenges:
- In two crystal prediction “blind tests” (tough benchmarks where the correct answer is hidden), PackFlow’s suggestions ended up closer to the experimental structures after relaxation (a polishing step) and achieved lower final energies than Genarris’s suggestions.
- With just 100 samples, PackFlow often got within a few kJ/mol of the experimental energy—a typical target for good crystal predictions.
Why It’s Important
- Saves time and effort: Getting good starting structures means scientists spend less time running expensive physics calculations to fix bad guesses.
- Improves accuracy: Better initial proposals lead to final results that match reality more often, which matters when choosing the right solid form for a drug or an electronic material.
- Practical and scalable: Because PackFlow outputs complete crystal structures (positions and unit cell), it plugs directly into standard physics tools used in materials science.
Implications and Impact
- Drug design: More reliable crystal predictions can help avoid issues like poor solubility or unexpected changes in drug stability.
- Organic electronics: Smarter packing leads to better performance in devices like OLED displays and solar cells.
- Research pipelines: PackFlow acts like a “proposal engine” that feeds high-quality candidates into existing workflows, cutting down cost and speeding up discovery.
The authors note that they focused on single-component molecular crystals (one type of molecule per crystal) and that future work could extend to more complex crystals (like co-crystals or solvates). Still, PackFlow’s approach—learning good packings, then aligning them with physics—offers a powerful and practical step forward in crystal structure prediction.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a concise, actionable list of what remains missing, uncertain, or unexplored in the work.
- All-atom generation is not addressed: hydrogens are added post hoc and PA uses heavy-atom-only rewards; quantify the impact on hydrogen-bond networks and explore joint heavy+hydrogen generation and alignment.
- Dependence on a single MLIP for relaxation and PA rewards is untested; assess robustness across multiple MLIPs and validate transferability to DFT-level relax-and-rank, including rank correlation and absolute energy errors.
- DFT validation is absent for the presented case studies; benchmark end-to-end outcomes (energies, structural matches) against dispersion-corrected DFT to confirm MLIP-driven gains persist at higher accuracy.
- Multicomponent crystals (co-crystals, salts, solvates) are excluded; extend conditioning and training to variable stoichiometries, charge states, proton transfer, and solvent inclusion.
- Space group and Z/Z′ are not predicted or enforced; develop mechanisms to predict/condition on space group and asymmetric-unit multiplicity, or to sample/score over them efficiently.
- No explicit symmetry constraints are used; investigate symmetry-aware architectures (e.g., Wyckoff/site priors, space-group equivariance) and measure effects on validity and efficiency.
- Treatment of conformational flexibility is implicit; evaluate torsional diversity and coverage, and integrate conformer priors or learned torsion models to better cover intramolecular basins.
- The PA “single-time” likelihood surrogate is an approximation; quantify its bias vs. exact path-based likelihood/KL estimators and its effect on stability and sample quality.
- Potential diversity collapse under PA is not analyzed; measure uniqueness, entropy, mode coverage, and density/motif spread pre/post-alignment and explore diversity-preserving regularizers.
- Only generation wall-clock is reported; quantify full pipeline cost (generation+hydrogenation+relaxations) and net savings vs. seeded heuristics at fixed success/recall targets.
- Evaluation breadth is limited (two blind-test cases, 100 samples each); conduct larger-scale, prospective CSP benchmarks using standard success metrics (e.g., rank of experimental form, RMSD15, energy window).
- No head-to-head comparisons with other generative CSP models (e.g., OXtal) are provided; perform controlled comparisons under identical relax-and-rank protocols.
- Conditioning on crystallization conditions (temperature, pressure, solvent) is absent; extend to conditional generation for polymorph control and test condition-specific recall.
- The mapping from input molecule to unit-cell content is unclear; learn or predict the number of molecules per cell/asymmetric unit and evaluate performance when Z/Z′ is unknown a priori.
- Thermodynamic effects (free energies, entropy/phonons) are not considered; test whether PackFlow proposals that are low in lattice energy remain favorable under finite-temperature free energy.
- Out-of-distribution robustness is noted but not quantified; design systematic OOD tests (new chemistries, size ranges, functional groups) and calibrate uncertainty and abstention strategies.
- Architectural equivariance is not enforced; evaluate whether SE(3)-equivariant backbones improve data efficiency, stability, and OOD performance relative to the current transformer.
- Lattice parameterization/constraints are approximate (unconstrained angles); assess alternative parameterizations (metric tensor/Cholesky) and enforce positive-definite cells during sampling.
- Independent coordinate/lattice time schedules show empirical gains; study learned/coupled schedules or curricula and provide theoretical/empirical analysis of why they help.
- Canonical unwrapping and centering may introduce biases; test augmentation over origins/cell choices and alternative unwrapping to ensure invariance and avoid leakage.
- Proxy-reward fidelity gap: heavy-atom rewards may misalign with all-atom relaxed outcomes; quantify this gap and explore lightweight all-atom proxy relaxations during PA.
- MLIP biases (e.g., hydrogen bonding, dispersion) may be exploited by PA; incorporate uncertainty-aware or committee-based rewards and periodic DFT-in-the-loop checks to curb reward hacking.
- Only ODE sampling is used; compare SDE-based samplers for exploration/diversity during PA and study quality–speed trade-offs and effect on relaxed outcomes.
- Pre-training does not include explicit steric/short-range physics; test adding differentiable steric/overlap penalties or coarse potentials to reduce clashes without PA.
- Dataset scope and selection bias are not detailed; release data/code, report composition/filters, and assess how curation choices affect generalization and metric outcomes.
- Property-targeted CSP is unexplored; extend PA to multi-objective alignment (e.g., density windows, packing motifs, electronic proxies) while maintaining stability.
- Scaling with system size/unit-cell complexity is not characterized; benchmark memory/time scaling and performance across molecular size, Z′, and unit-cell volume.
- Uncertainty quantification is missing; develop calibrated confidence/novelty scores (ensembles, posterior diagnostics) to prioritize candidates for costly relax-and-rank.
- Downstream structural comparison relies on AMD/RDF; add standard crystallographic metrics (e.g., COMPACK/RMSD15) and report final polymorph identification statistics.
- No prospective experimental validation is presented; attempt synthesis/crystallization of PackFlow-predicted polymorphs to establish real-world utility.
Practical Applications
Overview
Based on the paper’s findings and methods, the practical applications below focus on how PackFlow—joint lattice-and-coordinate generative CSP with reinforcement learning-based physics alignment—can be deployed or extended to impact industry, academia, policy, and daily life. Each item includes sector links, likely tools/products/workflows, and key assumptions or dependencies.
Immediate Applications
These applications are deployable now with currently available tools and validated performance (e.g., ~0.1 s/proposal, large reductions in density error and clash rates, improved relaxed energies vs. heuristics).
- PackFlow as a drop-in proposal engine in CSP pipelines
- Sectors: healthcare (pharmaceuticals), materials/software (organic electronics, OLED/OFET small-molecule crystals), chemicals
- Tools/workflows: Replace heuristic seed generators (e.g., Genarris) with PackFlow; continue hydrogen addition + MLIP relaxation + lattice-energy ranking; integrate with MLIPs like MACE, AIMNet2; use CCDC tooling for downstream analyses
- Value: Up to 83% reduction in density error vs. Genarris; ~40% fewer atomic clashes after PA; proposals relax to lower-energy minima in blind tests with a few kJ/mol of experimental polymorphs; ~0.1 s per heavy-atom proposal on a single V100 GPU
- Assumptions/dependencies: MLIP quality and domain fit; the heavy-atom proxy (pre-H addition) correlates with final stability; trained primarily on homomolecular crystal data; OOD generalization may still need energetic refinements
- Rapid polymorph risk screening in preformulation and process development
- Sectors: healthcare (drug development), chemicals
- Tools/workflows: Early-stage packings from PackFlow; prioritize relaxations for low-energy basins; use AMD/RDF/density to triage candidates; integrate with QbD workflows to identify potential high-risk polymorphs
- Value: Reduced relax-and-rank budget; faster coverage of plausible low-energy regions; improved proximity to experimental structures
- Assumptions/dependencies: MLIP ranking fidelity near 1–5 kJ/mol energy differences; access to accurate property models for downstream risk assessment
- Accelerated crystal initialization for periodic DFT
- Sectors: academia (computational chemistry/materials), software/AI for science
- Tools/workflows: Use PackFlow or PackFlow-PA proposals as stable initial geometries to reduce DFT geometry optimization iterations and failures
- Value: Fewer failed relaxations and shorter optimizations due to better densities and lower clash rates (especially with PA)
- Assumptions/dependencies: DFT settings and dispersion corrections consistent with MLIP trends; transferability of PA-guided proposals to DFT landscapes
- OLED and organic semiconductor packing exploration
- Sectors: electronics (display, sensors), materials R&D
- Tools/workflows: Generate plausible packings for small-molecule emitters/hosts; couple with property predictors (charge mobility, exciton diffusion, aggregation tendencies); screen stability-energy trade-offs
- Value: Faster exploration of structure–property relationships tied to packing; potential improvements in lifetime and efficiency via polymorph selection
- Assumptions/dependencies: Property predictors calibrated for target chemistries; thin-film vs. bulk-crystal differences handled via appropriate models
- IP and patent strategy support for solid forms
- Sectors: healthcare (brand/generics), legal/policy
- Tools/workflows: Rapid enumeration of plausible crystal packings; evaluate near-degenerate polymorphs to inform claim breadth and freedom-to-operate assessments
- Value: More comprehensive solid-form landscapes at lower computational cost
- Assumptions/dependencies: Acceptance of ML-assisted evidence in legal/regulatory contexts; DFT confirmation may still be necessary
- Teaching and benchmarking in computational crystallography
- Sectors: academia/education
- Tools/workflows: Use PackFlow to illustrate CSP stages; apply robust metrics (AMD, RDF Wasserstein, density error, clash rate); demonstrate physics alignment as post-training to improve stability
- Value: Accessible, fast demonstrations of modern generative CSP with clear evaluation tooling
- Assumptions/dependencies: Availability of curated datasets; MLIP integration in teaching environments
- Methodological reuse: RL physics alignment for generative flows
- Sectors: software/AI for science
- Tools/workflows: Apply GRPO with advantage mixing to other flow-matching generative tasks (e.g., protein backbones, periodic structures) where multi-objective stability proxies exist
- Value: Scale-free advantage mixing avoids hand-tuning reward magnitudes; KL regularization maintains distribution fidelity while improving physical validity
- Assumptions/dependencies: Appropriate proxy rewards; single-time surrogate for likelihood/score sufficiently stable for the target domain
Long-Term Applications
These applications require further research, scaling, or development (e.g., hydrogen-explicit generation, multi-component crystals, kinetic/processing effects, broader validation).
- End-to-end CSP with hydrogen-explicit generation and symmetry-aware sampling
- Sectors: healthcare, materials
- Tools/workflows: Extend PackFlow from heavy atoms to all atoms; integrate explicit space-group symmetry handling and Wyckoff site constraints; co-design generative sampling with downstream relaxations
- Value: Fewer post-processing steps; tighter alignment to periodic constraints; potentially more accurate initial energies
- Assumptions/dependencies: Reliable hydrogen placement in generation; symmetry-aware architectures; robust training across diverse space groups
- Co-crystal, salt, and solvate prediction
- Sectors: pharmaceuticals, specialty chemicals
- Tools/workflows: Extend datasets, conditioning, and model architectures for multi-component systems; integrate stoichiometry, counterion/solvent chemistry, and solution thermodynamics
- Value: Major expansion of CSP scope to common industrial solid forms; improved pipeline coverage of practical formulations
- Assumptions/dependencies: Diverse training data; MLIP/DFT fidelity for multi-component interactions; solvent/process-aware reward functions
- Process-aware polymorph prediction (thermodynamics + kinetics + environment)
- Sectors: pharmaceuticals, chemicals, electronics
- Tools/workflows: Couple PackFlow with kinetic models, solvent/pH/temperature effects, and nucleation/growth predictors; RL alignment using process-derived rewards (e.g., metastable persistence)
- Value: Increased relevance to what crystallizes under manufacturing conditions; rational control of polymorph outcomes
- Assumptions/dependencies: Accurate kinetic and environment models; new proxy rewards that capture non-equilibrium behavior
- Autonomous closed-loop solid-form discovery and optimization
- Sectors: robotics/lab automation, materials discovery
- Tools/workflows: Integrate PackFlow proposals with robotic crystallization platforms; property measurements feed back to update policies; Bayesian optimization over molecule + packing + process conditions
- Value: Rapid discovery of desired polymorphs with targeted properties; reduced human-in-the-loop burden
- Assumptions/dependencies: Reliable measurement pipelines; seamless data orchestration; safe and interpretable exploration strategies
- Device-level design optimization via packing–property co-optimization
- Sectors: electronics (OLED, OFET), energy (organic photovoltaics)
- Tools/workflows: Jointly optimize generated packings with charge transport, exciton dynamics, thermal stability, and mechanical robustness models; multi-objective RL incorporating property rewards
- Value: Translational path from molecular design to device performance; improved efficiency and lifetime
- Assumptions/dependencies: High-fidelity property models at crystalline scale; alignment between bulk crystal predictions and thin-film microstructures
- CSP-as-a-service platforms
- Sectors: software, cloud, consulting
- Tools/workflows: Managed cloud microservices offering PackFlow generation + MLIP/DFT relaxation + ranking + reporting; APIs for enterprise integration
- Value: Scalable access to state-of-the-art CSP; standardized metrics and audit trails
- Assumptions/dependencies: Data governance/IP confidentiality; compute cost control; service reliability and validation
- Standardization and regulatory guidance for ML-assisted CSP
- Sectors: policy/regulatory, healthcare
- Tools/workflows: Best-practice documents (metrics, validation protocols), acceptance criteria for ML-driven proposals in filings; harmonization with GxP and ICH guidelines
- Value: Smoother regulatory pathways for solid-form decisions; clearer evidence standards
- Assumptions/dependencies: Community consensus; demonstrated reproducibility and traceability of ML pipelines
- Cross-domain extension to other periodic materials (MOFs, zeolites, inorganic crystals)
- Sectors: energy (gas storage/separation), catalysis, electronics
- Tools/workflows: Adapt joint lattice–coordinate flow matching and physics alignment for frameworks and inorganic crystals; integrate domain-specific constraints (coordination chemistry, symmetry)
- Value: Broader impact of generative CSP beyond organics; accelerated structure generation in materials discovery
- Assumptions/dependencies: Suitable datasets; domain-aware MLIPs/DFT; handling of stronger bonding and symmetry constraints
- Advanced RL alignment with DFT-level rewards and uncertainty quantification
- Sectors: academia, software/AI for science
- Tools/workflows: Use batched DFT or high-accuracy MLIPs for reward signals; incorporate uncertainty calibration; develop efficient surrogate likelihoods for continuous flows
- Value: Closer alignment to true energetics; improved confidence in rankings and proposals
- Assumptions/dependencies: Compute budget; scalable DFT/MLIP infrastructure; robust surrogate estimators
- Societal benefits via improved product stability and reliability
- Sectors: daily life (medicines, consumer electronics)
- Tools/workflows: Upstream use of PackFlow-informed CSP reduces shelf-life risks in drugs and increases reliability of display components
- Value: More consistent product performance and availability
- Assumptions/dependencies: Successful translation of improved CSP predictions into manufacturing practices; validated correlations between predicted and realized forms in production environments
Glossary
- Advantage mixing: A multi-objective RL technique that linearly combines standardized advantages (rather than raw rewards) to balance competing objectives. "Advantage mixing linearly interpolates advantages instead of rewards for multi-objective post-training."
- Average-minimum-distance (AMD): A periodic structure descriptor measuring closeness between point patterns (robust to origin and small perturbations). "we use the average-minimum-distance (AMD) descriptor distance"
- Cambridge Crystallographic Data Centre (CCDC): The organization that runs the community CSP Blind Tests used to benchmark methods. "the CSP Blind Tests organized by the Cambridge Crystallographic Data Centre (CCDC)"
- Clash rate: The fraction of generated structures exhibiting severe short-range atomic overlaps under periodicity, used as a physical validity metric. "Clash rate quantifies severe short-range heavy-atom overlaps under periodic boundary conditions using a covalent-radius-based threshold."
- Covalent-radius-based threshold: A cutoff derived from covalent radii to detect unphysical interatomic overlaps. "using a covalent-radius-based threshold"
- Crystal structure prediction (CSP): The task of predicting stable crystal packings (and polymorphs) from molecular information. "Molecular crystal structure prediction (CSP) seeks to infer experimentally realizable crystal packings"
- Density functional theory (DFT): A quantum-mechanical method for accurate energetics used to rank crystal structures. "density functional theory (DFT)"
- Dispersion-corrected density functional theory (DFT): DFT augmented with dispersion interactions to better rank molecular crystals. "traditionally dispersion-corrected density functional theory (DFT) for final ranking"
- Flow matching: A generative training objective that learns a vector field to transport noise to data via an ODE. "PackFlow minimizes a flow-matching regression loss"
- Group relative policy optimization (GRPO): An RL algorithm that optimizes policies using within-group, relative advantages. "We post-train the pre-trained generator PackFlow-Base with group relative policy optimization (GRPO)"
- Heavy atom: Any non-hydrogen atom; used here as the focus of generation and scoring before adding hydrogens. "heavy-atom crystal proposals"
- Hydrogen-only relaxation: A brief optimization step allowing only hydrogen positions to relax before full relaxation. "a short hydrogen-only relaxation"
- Lattice energy: The energy of a crystal structure under periodic conditions, used for ranking polymorph stability. "lattice-energy ranking"
- Lattice parameters: The six unit-cell degrees of freedom (lengths and angles) defining the crystal lattice. "lattice parameters"
- Machine-learned interatomic potential (MLIP): A learned model that approximates energies and forces at near-DFT accuracy but lower cost. "machine-learned interatomic potentials (MLIPs) have begun to close the accuracy--cost gap"
- Optimal-transport (OT) interpolation schedule: A linear noise–data interpolation scheme used to construct training states for flow matching. "Using the optimal-transport (OT) interpolation schedule"
- Periodic boundary conditions: Modeling assumption that replicates the unit cell to represent an infinite crystal. "periodic boundary conditions"
- Polymorph: A distinct crystal packing of the same molecule, often with different properties and stability. "polymorphs can exhibit meaningfully different solubility, stability"
- Radial distribution function (RDF): A histogram of pairwise atomic distances used to assess packing statistics. "radial distribution function (RDF) histograms"
- Relax-and-rank: A CSP pipeline step: relax generated structures and rank them by energy. "relax-and-rank bottleneck"
- Space-group symmetry: The set of symmetry operations defining a crystal’s periodic symmetry. "space-group symmetry"
- Steric constraints: Physical limitations due to atomic sizes that prevent atoms from getting too close. "low clash rates indicate respect for steric constraints"
- Stochastic interpolants: A class of generative modeling techniques related to stochastic trajectories between noise and data. "stochastic interpolants"
- Time-dependent vector field: The learned field that guides denoising dynamics over time in flow matching. "PackFlow learns a time-dependent vector field"
- Unit cell: The fundamental repeating volume of a crystal that tiles space under periodicity. "within the unit cell"
- Unwrapped representation: A preprocessing where molecules are made whole across periodic boundaries to avoid discontinuous bonds. "Using an unwrapped representation was found to be an essential design choice"
- Wasserstein distance: A distributional metric used here to compare generated and experimental RDFs. "Wasserstein distances between radial distribution function (RDF) histograms"
- Wyckoff site: A set of symmetry-equivalent positions in a space group; molecules occupy orbits generated from these sites. "the symmetry mates of a Wyckoff site"
Collections
Sign up for free to add this paper to one or more collections.