- The paper introduces a novel computational framework that integrates Fisher Information Matrix, Hessian perturbation, eigenvalue decomposition, and Schur complement to establish scaling laws for parameter identifiability.
- It validates the framework through polynomial fitting benchmarks and applications in HIV dynamics and amyloid-β propagation, demonstrating precise uncertainty quantification.
- The results provide formal guarantees for improved model inference, enhanced practical identifiability, and robust uncertainty control in complex biological systems.
Scaling Laws for Parameter Identifiability and Uncertainty Quantification in Biological Data-Driven Modeling
Introduction: Mathematical Framework for Practical Identifiability
Integrating high-dimensional biological data into mechanistic modeling poses significant challenges in interpretability and generalizability, especially when parameter identifiability is compromised by the presence of ill-conditioned or flat likelihood landscapes. Traditional coordinate identifiability methods, primarily based on profile likelihoods, are computationally prohibitive and unreliable in the large-scale limit. This paper introduces a rigorous computational framework that synthesizes Fisher Information Matrix (FIM), perturbed Hessian analysis, eigenvalue decomposition (EVD), and Schur complement theory to reveal scaling laws governing practical identifiability across hierarchical orders. These laws provide a systematic taxonomy for parameter resolvability and uncertainty propagation in noisy, high-dimensional biological models.
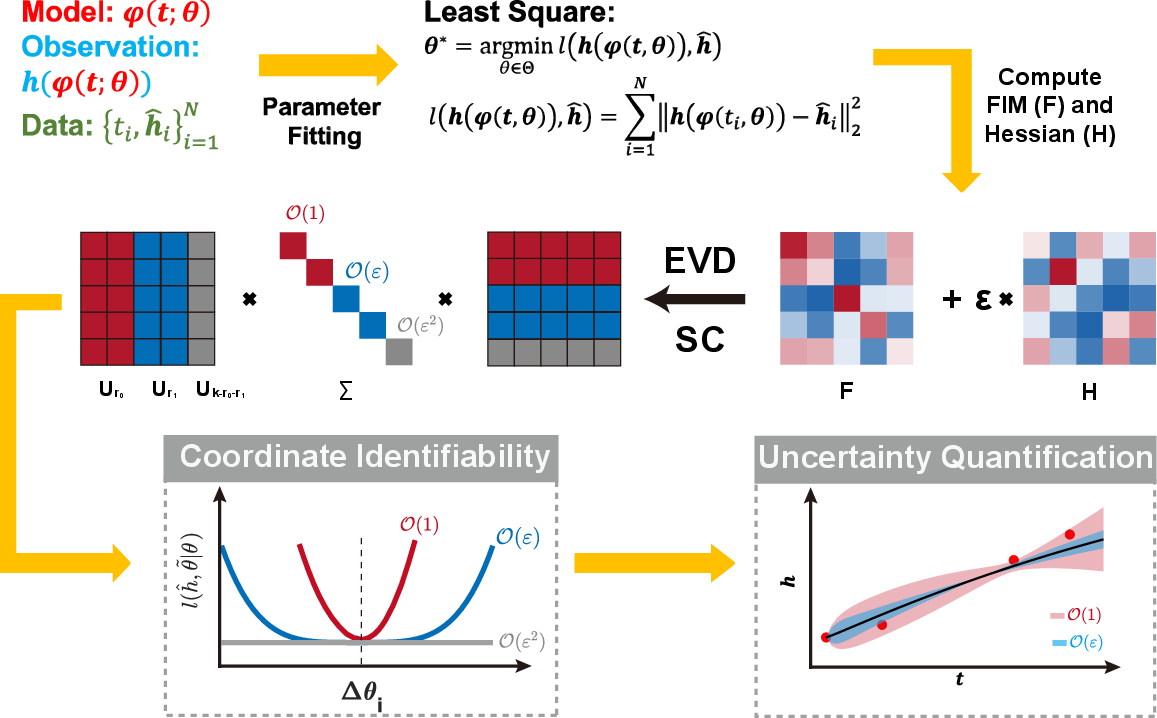
Figure 1: Higher-order parameter identifiability analysis: EVD and Schur complement classify parameters into zero-, first-, and second-order identifiable sectors; color codes mark eigenvalue-based order and non-identifiable subspaces.
Asymptotic Identifiability Theory: Hierarchical Metrics and Practical Criteria
The theoretical backbone of this framework is the asymptotic expansion of F+εH (FIM augmented by perturbation Hessian), enabling multi-order identifiability analysis. A hierarchy of identifiability metrics, Ki, is constructed: K0 recovers classical zero-order identifiability, while K1 and K2 resolve cases where the likelihood surface is flat (K0=0), including regions typically misclassified as non-identifiable by conventional techniques. Detailed mathematical theorems and corollaries establish that the existence of structurally informative directions in the null space of FIM (as modulated by H) determines higher-order practical identifiability, and link identifiability orders directly to uncertainty containment principles.
Numerical Benchmark: Polynomial Fitting and Higher-Order Identifiability
A polynomial fitting benchmark is employed to establish empirical congruence between the novel Ki metrics and classical profile likelihood analysis. Notably, parameters with flat likelihood surfaces are robustly classified as first-order identifiable (K1>0), avoiding false negatives that arise when only zero-order criteria (K0) are used. The eigenvalue and eigenvector decomposition elucidates the spectral hierarchy of parameter resolvability, and uncertainty quantification (UQ) demonstrates that confidence intervals over non-identifiable subspaces are nested according to identifiability order.
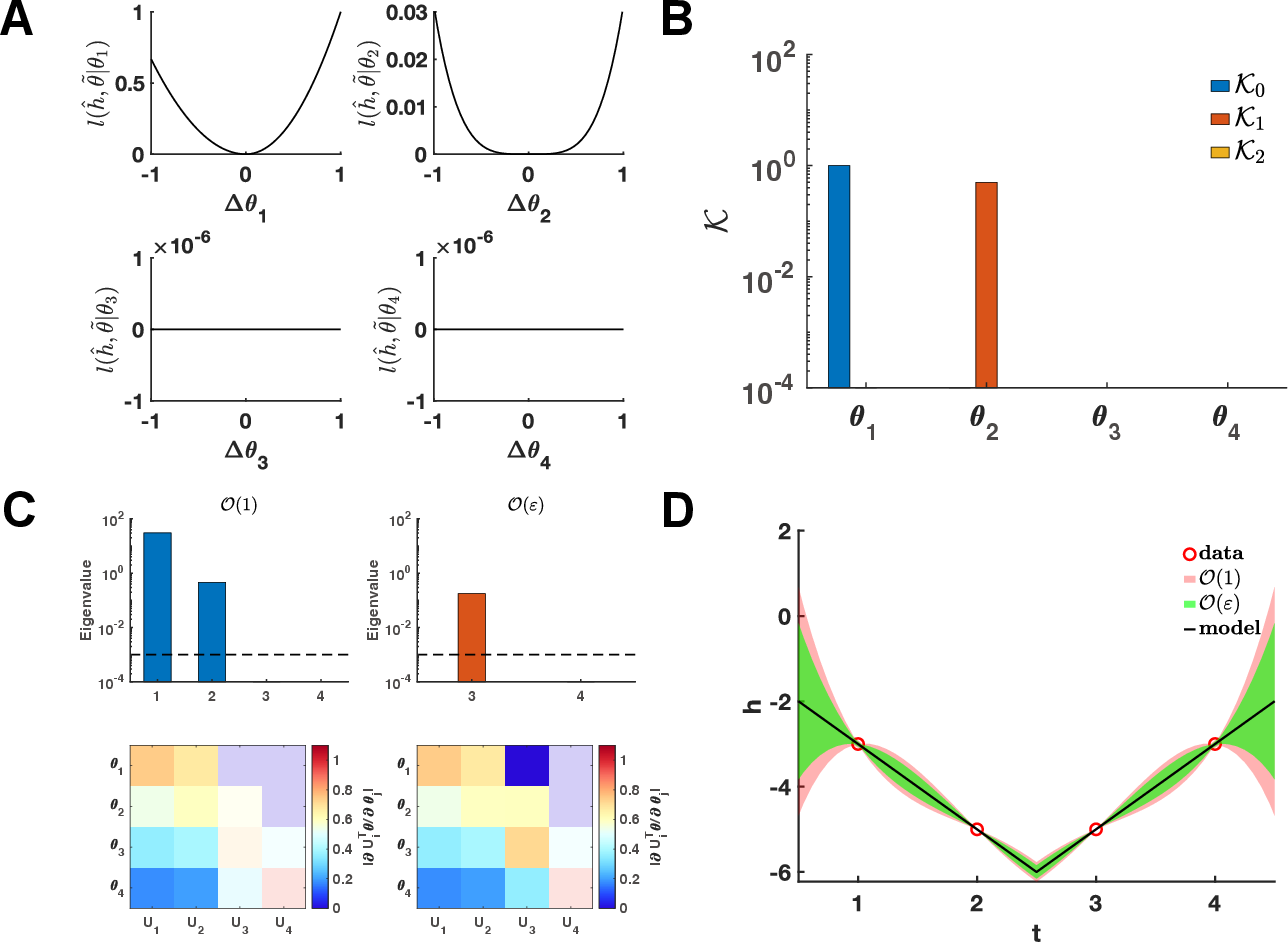
Figure 2: Validation of polynomial model identifiability: Ki metrics align with profile likelihood; uncertainty intervals shrink at data points, confirming local invariance on the non-identifiable manifold.
Application to HIV Host-Virus Dynamics: Identifiability Architecture and UQ Mapping
The scaling law framework is applied to a canonical HIV infection ODE model, with six biological parameters governing plasma viral load dynamics. Eigen-coordinates are classified into zero-order (robustly identifiable), first-order (contingent on higher-order perturbation), and non-identifiable sectors—precisely mapping onto specific biological mechanisms such as virion production and clearance rates. UQ analysis reveals that non-identifiable parameters exert broad influence during initial outbreak and relapse phases, whereas first-order non-identifiable parameters modulate uncertainty primarily in early infection windows, validating the utility of hierarchical identifiability for temporal decomposition of predictive uncertainty.
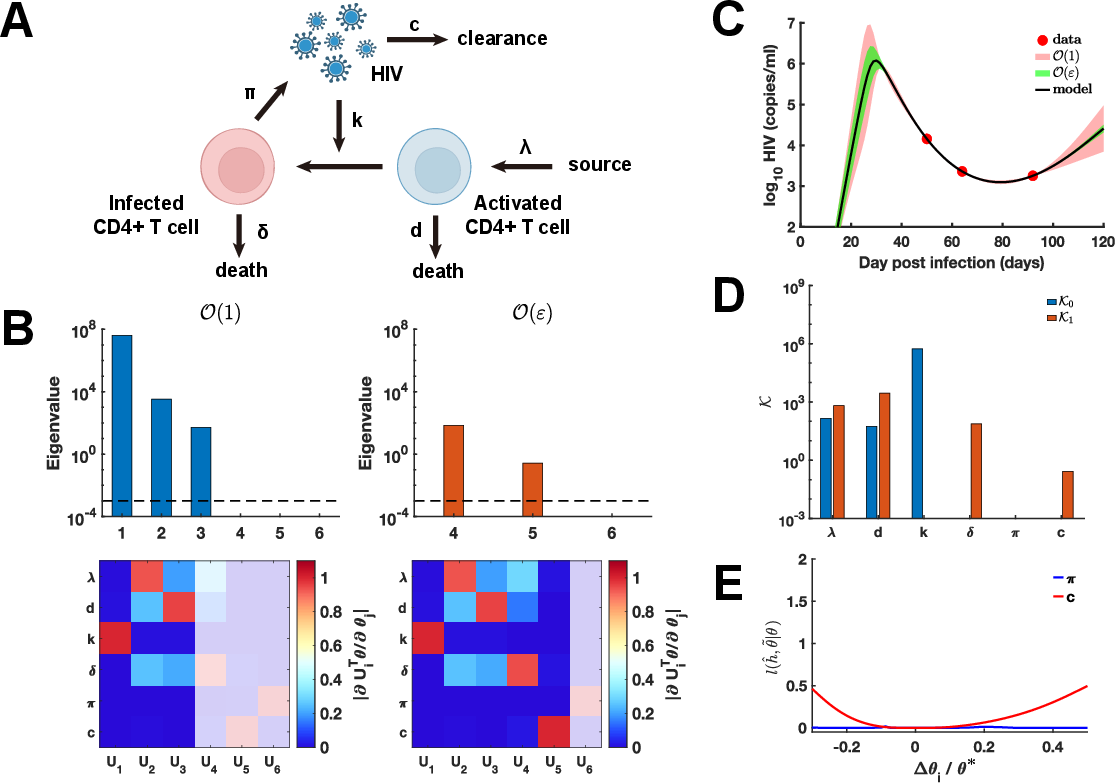
Figure 3: Higher-order identifiability in HIV ODE: Eigenvalue heatmap, UQ showing phase-specific uncertainty, and coordinate-wise profile likelihood confirmation for c and π.
Spatiotemporal Amyloid-β Propagation: High-Dimensional Hierarchical Scaling
Extending to networked PDE modeling of amyloid-β (Aβ) dynamics across 68 cortical regions, the identifiability framework reveals a complex spectral hierarchy in parameter space, with both zero-order and first-order identifiable coordinates dominant. Regional production rates exhibit robust identifiability; carrying capacities are either first-order identifiable or non-identifiable. The uncertainty structure, as quantified by UQ, is highly order-dependent. Zero-order UQ captures terminal states; first-order UQ resolves rapid accumulation phases, pinpointing windows critical for disease progression diagnosis.
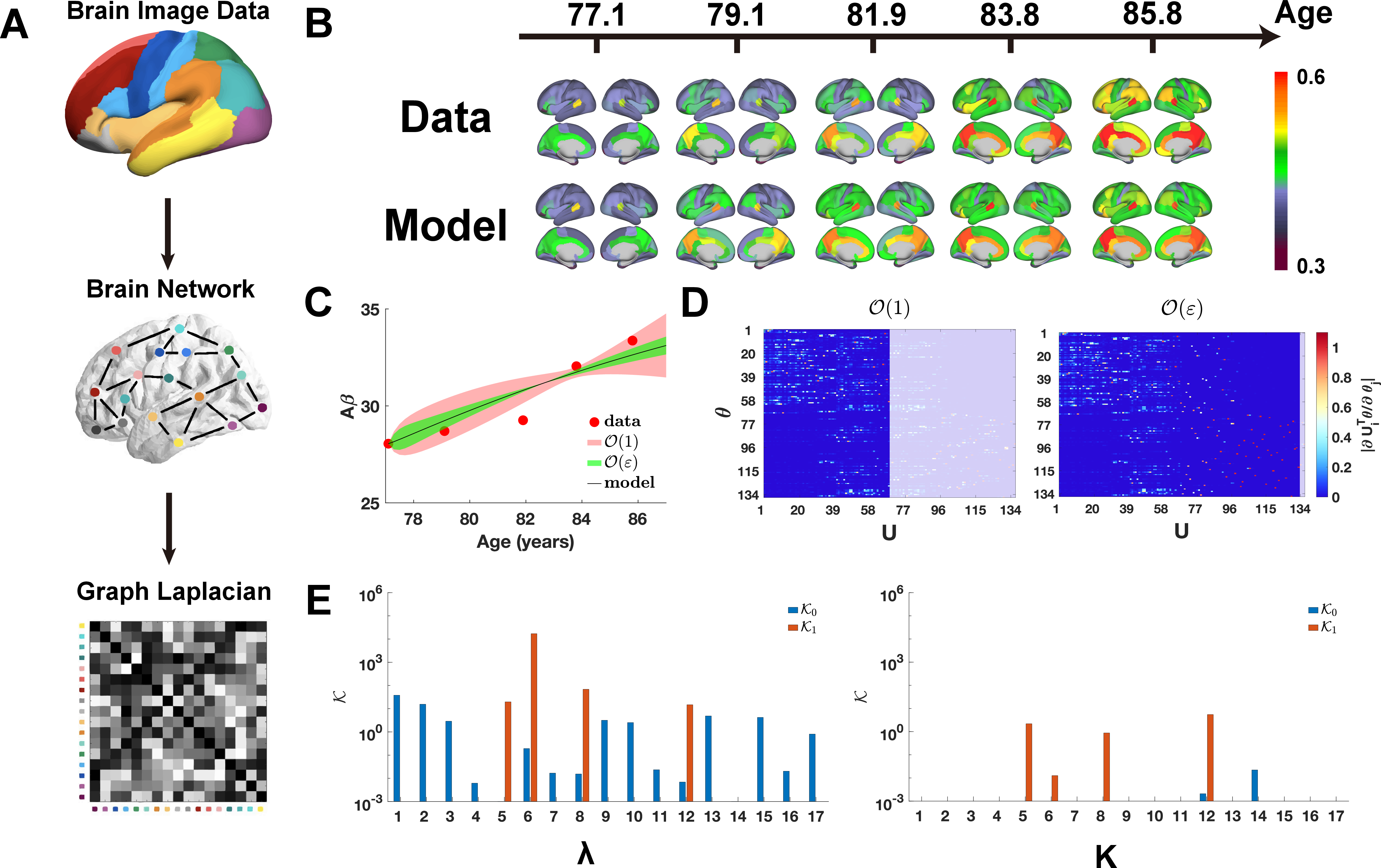
Figure 4: Identifiability hierarchy for Aβ propagation: Image pipeline, model reconstruction, and UQ revealing age-dependent uncertainty suppression in supra-threshold orders.
Theoretical and Practical Implications
The results have direct implications for data-driven biological modeling at every scale. The hierarchical identifiability analysis provides formal guarantees for parameter resolvability, enabling efficient diagnosis and preventing misleading inferences due to numerical artifacts or data sparsity. The associated UQ protocol rigorously quantifies the extent of epistemic uncertainty, ensuring confidence intervals are appropriately bounded relative to the order of identifiability. The framework advances the field by replacing exhaustive likelihood scanning with analytical diagnostics that scale to large mechanistic digital twins and complex biological networks.
Conclusion
This work introduces scaling laws for parameter identifiability and uncertainty quantification grounded in asymptotic expansion and spectral theory. By integrating hierarchical metrics, eigen-structure, and Schur complement analysis, it bridges practical inference with theoretical guarantees, enhancing reliability and interpretability in biological model fitting. The framework is validated numerically and in real-world systems—demonstrating its scalability, analytic rigor, and utility for precision medicine, mechanistic digital twin construction, and large-scale biological inference. Future developments will likely focus on extending the framework to nonlinear loss functions, generalized objective regimes, and hybrid data modalities, as well as fostering automatic identifiability diagnostics for next-generation AI-powered biological modeling.