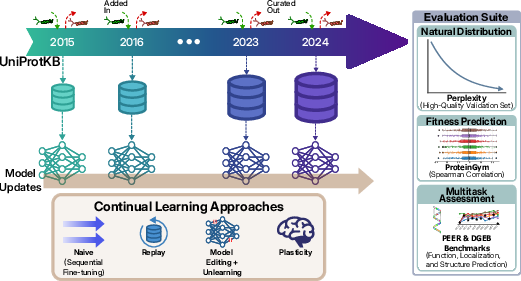
CoPeP: Benchmarking Continual Pretraining for Protein Language Models
Abstract: Protein LLMs (pLMs) have recently gained significant attention for their ability to uncover relationships between sequence, structure, and function from evolutionary statistics, thereby accelerating therapeutic drug discovery. These models learn from large protein databases that are continuously updated by the biology community and whose dynamic nature motivates the application of continual learning, not only to keep up with the ever-growing data, but also as an opportunity to take advantage of the temporal meta-information that is created during this process. As a result, we introduce the Continual Pretraining of Protein LLMs (CoPeP) benchmark, a novel benchmark for evaluating continual learning approaches on pLMs. Specifically, we curate a sequence of protein datasets derived from the UniProt Knowledgebase spanning a decade and define metrics to assess pLM performance across 31 protein understanding tasks. We evaluate several methods from the continual learning literature, including replay, unlearning, and plasticity-based methods, some of which have never been applied to models and data of this scale. Our findings reveal that incorporating temporal meta-information improves perplexity by up to 7% even when compared to training on data from all tasks jointly. Moreover, even at scale, several continual learning methods outperform naive continual pretraining. The CoPeP benchmark offers an exciting opportunity to study these methods at scale in an impactful real-world application.


Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
CoPeP: A simple guide for teens
What this paper is about (the big idea)
Proteins are tiny machines that keep our bodies (and all living things) running. Each protein is made from a chain of amino acids—like letters forming words and sentences. Scientists now use “protein LLMs” (kind of like the tools that learn human language) to read protein sequences and guess what they do.
But there’s a problem: the main protein database (UniProt) is always changing—new proteins get added, and some get corrected or removed. Instead of training a new model from scratch every time, this paper explores how to keep updating a model as new data arrives, like keeping a phone’s operating system up to date. The authors build a new benchmark, called CoPeP, to test different ways of doing this “continual learning” for protein models.
What questions did the researchers ask?
- Can we keep improving a protein model by training it year after year as the database grows, without starting over?
- Does using the “history” of the database (which sequences stayed for years vs. which were later removed) help the model learn better?
- Which continual learning strategies work best for different kinds of protein tasks?
How they studied it (in everyday language)
Think of UniProt as a huge, ever-growing library of protein “sentences.” The authors:
- Collected 10 yearly “snapshots” of this library from 2015 to 2024 (so: 2015 data, then 2016, and so on).
- Trained a protein LLM step by step through the years, like teaching a student a little more each grade, rather than starting over.
- Used the database’s history as a clue: sequences that stick around for years are probably real and useful; those that get removed later might be wrong or less helpful.
- Compared several update strategies:
- Plain sequential training: just keep training on each new year’s data.
- Replay: mix in older, trustworthy examples (especially ones that persist over many years) while learning new ones—like revising old lessons while learning new topics.
- Plasticity helpers (to keep the model “flexible” and able to learn new things):
- Shrink and Perturb: nudge the model’s “memory” slightly and add a tiny bit of noise so it doesn’t get stuck.
- Hare and Tortoise: keep two versions of the model (fast-learning and slow, stable) and regularly sync them.
- Unlearning (teaching the model to forget outdated or wrong info):
- Gradient Ascent: actively “pushes away” what the model learned from now-removed data.
- Random Labels: scrambles the targets for removed data so the model stops trusting them.
- Tested the models on:
- A high-quality validation set of trusted proteins (to see how well the model understands natural protein sequences).
- ProteinGym (predicting how mutations affect protein fitness—important for protein design).
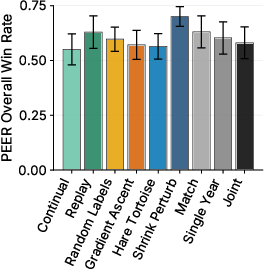
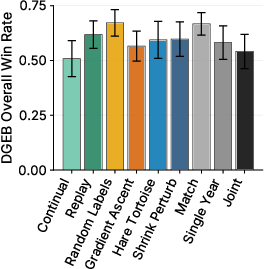
- Two big multi-task collections (PEER and DGEB) covering many real-world protein questions (like function, location, and relationships). In total, they evaluate performance across more than 30 tasks.
Helpful translation of a few terms:
- “Perplexity”: a score for “how surprised the model is” by real protein sequences. Lower is better (less surprise means it understands the “language” well).
- “Replay”: reviewing some old examples while learning new ones.
- “Plasticity”: how easily the model can still learn new things.
- “Unlearning”: helping the model forget information that turns out to be wrong or unhelpful.
What they found (and why it matters)
Here are the main takeaways:
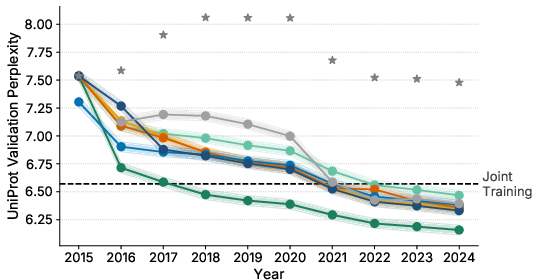
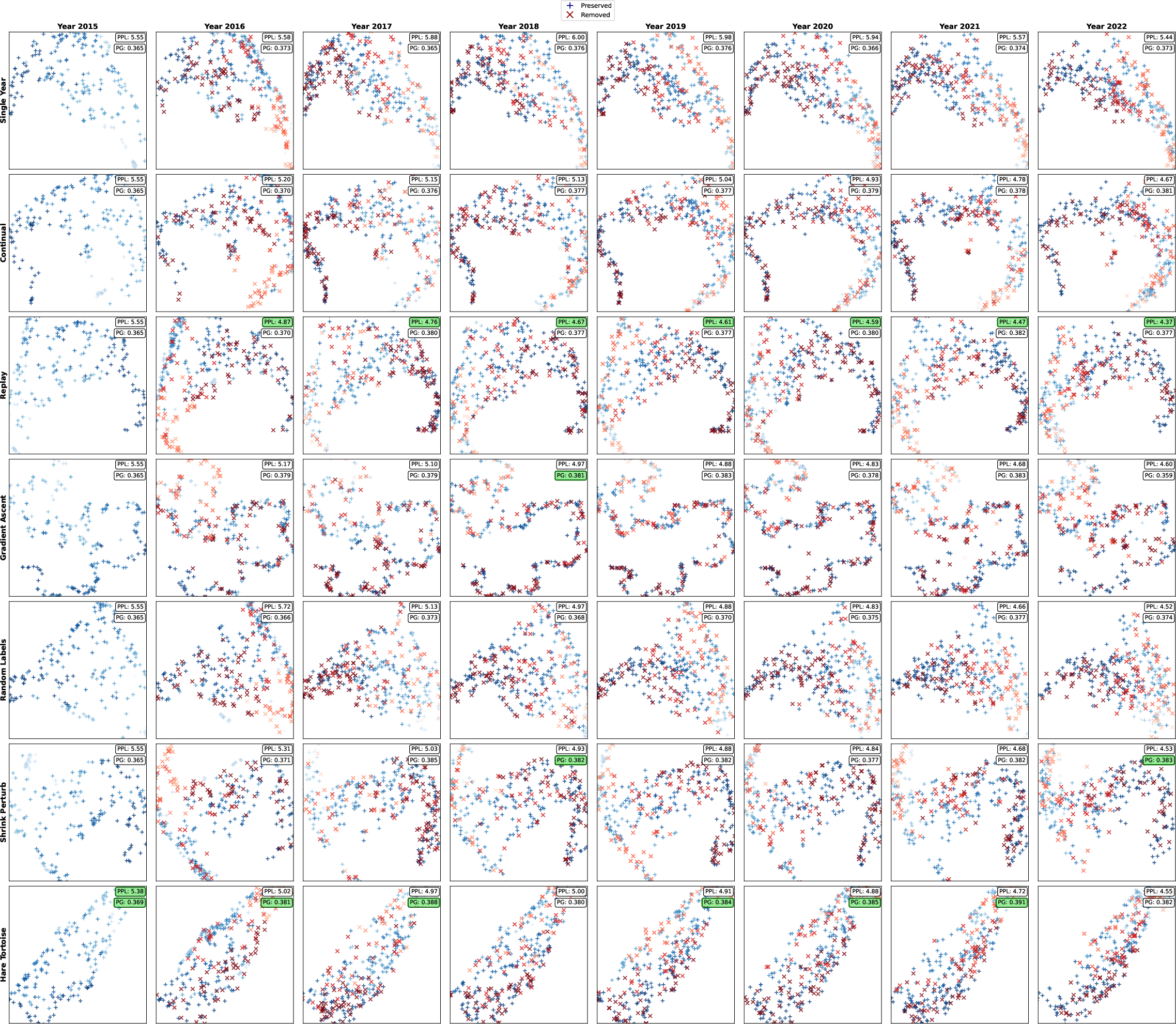
- Using the database’s history helps. Prioritizing sequences that persist over years improved the model’s perplexity by up to about 7%—even compared to training on all the data at once.
- Quality beats quantity (sometimes). Training only on sequences that appear in both two different years (so, more “trusted”) sometimes beat training on a larger, single-year dataset—even with less data.
- Continual learning works—and can beat “just retrain on everything.” Several update strategies beat both:
- the naïve approach (just keep training year by year without special tricks), and
- “joint training” (training once on the combined data from all years).
- Different methods shine on different tasks:
- Replay (favoring long-lasting sequences) did best on modeling natural protein distributions (lower perplexity on the high-quality validation set).
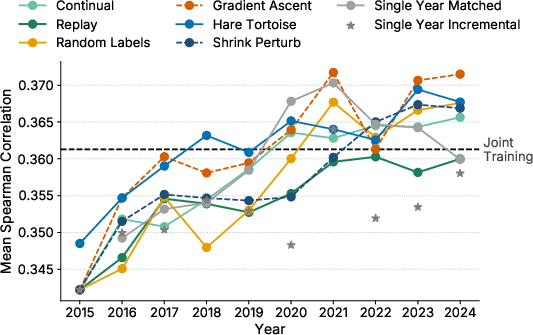
- Hare and Tortoise and Gradient Ascent (unlearning) did best on ProteinGym (predicting mutation effects).
- Shrink and Perturb and Random Labels did best on the multi-task benchmarks (PEER and DGEB), which focus on general protein understanding and more diverse, less-studied sequences.
- Joint training (using everything, including sequences later removed from UniProt) can actually be worse—probably because it includes examples that later turned out to be poor-quality or not real proteins (like pseudogenes).
Why this matters: It shows we can keep protein models up to date efficiently and even improve them, which is important for discovering new medicines and understanding biology faster.
What this could change (the impact)
- Faster, cheaper updates: Instead of starting from scratch, labs can keep improving protein models as new data arrives.
- Smarter data use: Paying attention to what stays in the database over time helps filter out noise and improves learning.
- Better tools for science and medicine: Tasks like predicting mutation effects or understanding protein functions are essential for designing drugs and treatments. Matching the right update method to the right task can boost accuracy.
- A new testing ground for the community: CoPeP gives researchers a realistic, large-scale benchmark to develop and compare continual learning methods for biology.
In short: This paper shows that “learning over time” with smart strategies—and using the database’s history—can make protein models better, more reliable, and more useful for real-world science.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a consolidated list of specific gaps and open problems left unresolved by the paper that future researchers can address.
- Scalability to larger and different architectures:
- Do the reported trends (e.g., benefits of temporal metadata, effectiveness of Hare & Tortoise, Shrink & Perturb, Replay) hold for larger pLMs (e.g., ≥650M+ parameters) and for autoregressive architectures (e.g., ProGen2/GPT-style) rather than only a 120M bidirectional masked LM?
- Compute- and token-budget fairness:
- The joint-training baseline’s training budget is not clearly matched to the cumulative compute of continual methods. A controlled comparison under equal token budgets or equal wall-clock compute remains missing.
- Streaming protocol realism:
- CoPeP allows full access to all past data (an unbounded replay pool). How do methods fare under realistic constraints where only a memory-bounded buffer or no historical data can be stored?
- Replay memory design and costs:
- The unbounded, multiplicity-weighted replay is compute- and storage-intensive at 580M+ sequences. What are the trade-offs with bounded buffers (e.g., reservoir sampling, coresets, sketches) and what is the impact on performance?
- Replay sampling criteria beyond persistence:
- Multiplicity (temporal persistence) is the sole replay importance signal. Would other metadata (evidence codes, Swiss-Prot vs TrEMBL status, taxonomic diversity, functional coverage, length, cluster centrality) yield better outcomes or reduce bias?
- Bias induced by persistence weighting:
- Temporal persistence favors well-studied, long-lived entries and early-sequenced taxa. Does this entrench taxonomic and functional biases? Quantify shifts across domains of life, underrepresented clades, and rare functions.
- Causal interpretation of removal signals:
- Sequences removed between releases are treated as lower quality by implication. What fraction are removed for redundancy vs true errors (e.g., pseudogenes)? Can reason-specific weighting or exclusion yield better curation than a blanket “removed = noisy” assumption?
- Deduplication and decontamination practices:
- ProteinGym evaluations were not decontaminated against pretraining. How much do results change with strict wild-type decontamination and varying identity thresholds (e.g., 50%, 70%, 90%)?
- Generalization beyond UniRef100:
- Only UniRef100 cluster representatives are used. How do results change with UniRef90/50 (different redundancy/coverage trade-offs) or with raw UniProtKB (including within-cluster diversity)?
- Sequence length truncation:
- Training is capped at length 512. What is the impact on long proteins and on downstream tasks sensitive to long-range dependencies? Are longer-context models or curriculum strategies beneficial?
- Optimizer-state carryover and LR scheduling:
- The study adopts Warmup-Stable-Decay with resets between tasks, but the effect of carrying over vs resetting optimizer states (Adam moments) and alternative schedules (e.g., cosine restarts, per-token LR) is not systematically ablated.
- Task-agnostic vs task-aligned pretraining objectives:
- Perplexity gains did not consistently translate to ProteinGym. Would objective variants (e.g., span masking, masked distillation, contrastive objectives, evolutionary-aware objectives) better align with downstream fitness prediction?
- Combining orthogonal CL strategies:
- The paper suggests complementary strengths (e.g., replay for natural distribution, unlearning/plasticity for fitness prediction), but does not test combined or adaptive strategies (e.g., multi-objective schedules, alternating phases, controller policies).
- Evaluation breadth:
- Structural prediction tasks (e.g., contacts, tertiary structure) and ligand-aware tasks were excluded due to cost. What are the effects of continual pretraining on structure-aware tasks (even on reduced subsets or lighter proxies) and on structure–function integration?
- Downstream task stratification:
- Aggregate metrics mask heterogeneity. Which taxa, protein families, functions, or mutational contexts gain/lose with each method? Fine-grained per-assay/per-taxon analyses are needed to detect systematic strengths/weaknesses.
- Persistence-based filtering trade-offs:
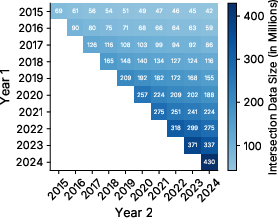
- Intersections across years improved perplexity with reduced data. What is the optimal persistence threshold (e.g., ≥2/≥3 years) and how does it trade off with coverage of novel/rare proteins and downstream performance?
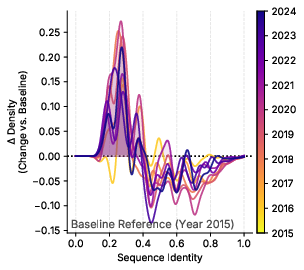
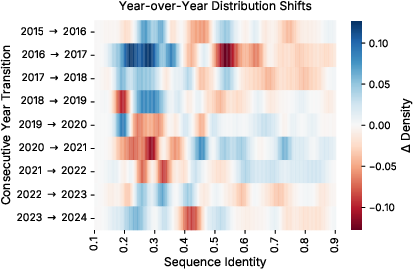
- Temporal-drift characterization:
- Drift is analyzed primarily via sequence identity to a fixed validation set. A richer analysis (taxonomy, domains/families, function ontology, length/AA composition, evidence code shifts) could inform targeted CL strategies.
- PEFT and modular continual learning:
- Parameter-efficient methods (adapters, LoRA), expandable architectures (e.g., MoE, progressive nets), and gating were not evaluated. Can these approaches reduce forgetting or improve plasticity at lower compute/memory?
- True unlearning guarantees:
- “Forget set” unlearning is proxy-based (gradient ascent, random labels) and not certified. How well do these methods remove information influence (e.g., via influence functions, membership inference) and how do they compare to exact retraining approximations at scale?
- Continual pretraining without full historical access:
- In settings with privacy/IP restrictions or streaming data without archival, can one achieve comparable performance using only deltas, sketches, or summary statistics of past data?
- Objective–metric mismatch:
- The paper shows that reduced perplexity doesn’t guarantee gains on ProteinGym. Can one design pretraining-time multi-task or auxiliary objectives that better predict (and improve) zero-shot mutation ranking performance?
- Out-of-distribution and metagenomic generalization:
- How do methods perform on truly novel, recent metagenomic proteins or low-identity sequences not present in historical releases? Include “future-year” held-out sets to measure forward-transfer.
- Contamination in PEER/DGEB:
- While fine-tuning splits exist, potential leakage from pretraining corpora to test sets is not assessed. A systematic pretraining decontamination study for PEER/DGEB is missing.
- Compute/energy and reproducibility details:
- Full training cost, token counts per method, and seeds/variance across all benchmarks are not exhaustively reported. Robust significance testing across runs would strengthen conclusions.
- Alternative validation sets:
- The validation set skews toward well-studied proteomes. Include complementary validation sets emphasizing underrepresented taxa, environmental samples, and recent discoveries to avoid overfitting to “early” proteomes.
- Temporal metadata beyond multiplicity:
- Age (time since first seen), survival curves, re-annotations, review status changes, and curator notes are unexploited. Can richer temporal signals improve filtering/replay over multiplicity alone?
- Practical deployment guidance:
- The paper shows task-dependent trade-offs but does not provide procedures for selecting or adapting methods during deployment (e.g., automatic method switching based on validation signals relevant to the intended downstream use).
- Benchmark extension:
- Only a decade (10 tasks) is considered. How do methods behave over longer horizons (e.g., 20–30 tasks) and with finer-grained temporal slices (e.g., quarterly releases) where shifts may be subtler but more frequent?
Practical Applications
Immediate Applications
Below are actionable use cases that can be deployed now, leveraging the paper’s benchmark (CoPeP), methods (temporal replay, plasticity-preserving, unlearning), and empirical insights (temporal persistence as a data-quality signal).
- Continual update pipelines for protein LLMs (pLMs) to keep models current with UniProt/UniRef releases (Healthcare/Pharma/Biotech; Software)
- Workflow: Replace periodic “train-from-scratch” with continual pretraining using Warmup–Stable–Decay (WSD) and one of the validated strategies:
- Temporal Replay (multiplicity-weighted) if the goal is to better model the natural protein distribution and improve perplexity/sequence recovery.
- Hare & Tortoise or Gradient Ascent if the goal is stronger zero-shot mutational effect prediction (ProteinGym-like use).
- Shrink & Perturb or Random Labels if the goal is better transfer to diverse downstream tasks (PEER/DGEB).
- Tools/products: “Protein Model Update Service” that ingests each UniRef100 release, applies the selected method, and publishes updated checkpoints/embeddings via an internal model hub/API.
- Assumptions/dependencies: Access to recent UniRef releases; compatibility with the organization’s base model (scaling from ~120M to larger models may require tuning); clear target task to guide method choice.
- Temporal persistence-based data curation to improve pretraining corpora (Healthcare/Pharma/Biotech; Academia; Data infrastructure)
- Workflow: Use sequence “multiplicity” (count of consecutive years present) and/or set intersections across releases to filter or reweight training examples. This exploits the paper’s finding that temporal persistence correlates with improved modeling (e.g., up to ~2.5% perplexity gain at smaller dataset sizes and up to ~7% vs joint training in some settings).
- Tools/products: A “Temporal Filter” dataset preprocessor that computes per-sequence multiplicity, filters transient entries, and emits weighted sampling indices.
- Assumptions/dependencies: Temporal presence is a proxy for data quality but not ground truth; risk of bias toward well-studied proteomes; must align filtering with downstream objectives.
- Cost- and carbon-efficient model maintenance via continual pretraining (Energy/Sustainability; Software/IT)
- Workflow: Swap full retraining for continual pretraining checkpoints and WSD scheduling; maintain persistent replay buffers and task-specific unlearning/EMA procedures to avoid performance regressions.
- Tools/products: Sustainability dashboards tracking compute saved and CO₂e reductions by moving from scratch retraining to continual pretraining.
- Assumptions/dependencies: Stable MLOps pipeline; availability of archival checkpoints; validation that continual checkpoints meet current accuracy/robustness bars.
- “Unlearning” to align models with evolving curation decisions and compliance needs (Healthcare/Pharma; Policy/Compliance; Biosecurity)
- Workflow: Apply Gradient Ascent or Random Labels to actively remove influence of sequences that UniProt curates out (e.g., pseudogenes, redundancies, contaminated entries), keeping the foundation model aligned with trusted corpora.
- Tools/products: “Unlearning Module” integrated into the update pipeline to handle forget sets (e.g., prior-year entries not present in the new release).
- Assumptions/dependencies: Clear definition of the forget set; guardrails to prevent over-unlearning; monitoring to ensure downstream performance isn’t unduly harmed.
- Rapid, task-optimized model selection for downstream R&D pipelines (Healthcare/Pharma/Biotech)
- Workflow: Maintain multiple updated variants optimized for different tasks:
- Distribution modeling/annotation: Temporal Replay variant.
- Zero-shot mutation effect prediction: Hare & Tortoise or Gradient Ascent variant.
- Broad representation learning for fine-tuning: Shrink & Perturb or Random Labels variant.
- Tools/products: An internal “model matrix” catalogue linking benchmark results (UniProt validation, ProteinGym, PEER, DGEB) to model selection guidance for project teams.
- Assumptions/dependencies: Benchmark coverage must reflect intended downstream workloads; ongoing validation on in-house assays.
- Plug-and-play embedding refresh for bioinformatics pipelines (Healthcare/Pharma/Biotech; Software)
- Workflow: Periodically update protein embeddings used for variant effect prediction, protein annotation, function/localization prediction, and retrieval with the latest continual-pretrained pLM.
- Tools/products: Embedding service/API with versioned endpoints; automatic backfills in data lakes; downstream retraining triggers.
- Assumptions/dependencies: Controlled versioning; stability commitments for production services; compatibility with downstream models’ feature schemas.
- Open benchmarking and method evaluation using CoPeP (Academia; Open-source; Software)
- Workflow: Use the released code, checkpoints, and datasets to evaluate new continual learning approaches at realistic scale and to reproduce baseline results.
- Tools/products: Academic courses, workshops, and tutorials that demonstrate temporal replay and unlearning on CoPeP; CI pipelines for automated benchmarking.
- Assumptions/dependencies: Access to sufficient compute; careful comparison against baselines due to data shift nuances.
- Data QA and triage for public or internal protein databases (Academia; Data infrastructure)
- Workflow: Use temporal multiplicity and model perplexity changes to flag transient or suspicious entries for curation attention.
- Tools/products: “Temporal QA Dashboard” that visualizes persistence patterns and model fit changes across releases.
- Assumptions/dependencies: Curator bandwidth; policies for how to act on flagged entries; acceptance that model fit is an indirect signal.
- Institutional policies for model maintenance and sustainability (Policy; Research IT)
- Workflow: Adopt internal guidelines prioritizing continual pretraining over full retraining when feasible; require model update audit trails (releases used, unlearning runs, and replay settings).
- Tools/products: Standard operating procedures (SOPs) for foundation-model upkeep in life sciences.
- Assumptions/dependencies: Organizational buy-in; compliance alignment; traceable data provenance.
Long-Term Applications
The following opportunities will require additional research, scaling, integration, or validation before widespread deployment.
- Streaming/online model updating with frequent releases and mixed data sources (Healthcare/Pharma/Biotech; Software)
- Vision: Move from annual batch updates to monthly or continuous updates that combine UniProt releases with in-house sequences and metagenomic data, handling non-stationary drift in near real-time.
- Potential product: “Real-time Biological Foundation Model Service” with rolling replay buffers and automated unlearning of deprecated entries.
- Assumptions/dependencies: Robust drift detection, scheduling, and rollback; scalable unbounded replay or approximate alternatives; monitoring for stability vs plasticity.
- Closed-loop wet-lab integration and active learning (Healthcare/Pharma/Biotech)
- Vision: Use temporally-aware pLMs to prioritize variants/proteins for experimental assays; feed results back to the model via continual pretraining or fine-tuning to improve design cycles.
- Potential workflow: Design–Make–Test–Learn pipelines that exploit multiplicity-weighted priors and task-specific continual strategies.
- Assumptions/dependencies: Data-sharing across teams; standardized assay readouts; mechanisms to avoid feedback loops that reinforce dataset biases.
- Federated and privacy-preserving continual pretraining across institutions (Healthcare/Pharma; Academia; Policy)
- Vision: Institutions contribute updates via federated replay and controlled unlearning to maintain a shared model without exposing raw sequences.
- Potential product: “Federated CoPeP” with secure aggregation, replay scheduling, and audit trails for forget sets.
- Assumptions/dependencies: Privacy regulations, legal agreements, and robust federated optimization under heterogeneous distributions.
- Regulatory frameworks for biological foundation model maintenance (Policy/Regulatory)
- Vision: Codify model update, replay, and unlearning procedures for audited traceability in regulated settings (e.g., clinical decision support or safety review for generative design tools).
- Potential tools: Model maintenance logs, unlearning certificates, and dataset-versioning registries.
- Assumptions/dependencies: Multi-stakeholder consensus; standards for documenting and validating model modifications.
- Cross-domain generalization of temporal-meta learning (Software; Energy; Finance; Education)
- Vision: Apply temporal persistence weighting and multiplicity-based replay to other evolving corpora (e.g., chemical LLMs, clinical coding, legal/financial text).
- Potential products: “Temporal Data Engine” libraries that add multiplicity-aware sampling and unlearning to any sequence model pipeline.
- Assumptions/dependencies: Comparable temporal metadata quality; validation that persistence correlates with data reliability in the target domain.
- Safety and biosecurity guardrails via unlearning (Policy; Biosecurity; Healthcare)
- Vision: Proactively unlearn or downweight sequences associated with toxins/pathogenicity or entries later deemed unsafe or erroneous, and log the process for audit.
- Potential tools: “Safety Unlearning” modules linked to curated blocklists and risk signals.
- Assumptions/dependencies: Reliable risk annotations; careful evaluation to avoid catastrophic forgetting of benign regions affecting utility.
- Clinical-grade variant effect prediction and diagnostics (Healthcare)
- Vision: Leverage stronger pLMs to support clinical interpretation of variants through improved embeddings and zero-shot fitness prediction pipelines.
- Potential products: Decision-support tools integrating updated pLM scores into variant pathogenicity assessments.
- Assumptions/dependencies: Extensive clinical validation, external benchmarks representative of patient populations, regulatory approvals, and robust uncertainty estimates.
- Large-model scaling and multi-objective continual pretraining (Software/AI; Healthcare)
- Vision: Scale beyond 120M parameters with method ensembles (e.g., combining replay with plasticity preservation and targeted unlearning) to meet diverse downstream objectives simultaneously.
- Potential tools: Orchestrators that schedule per-task interventions (replay vs EMA resets vs unlearning) based on live evaluation metrics.
- Assumptions/dependencies: Compute budgets; stability of method combinations; principled selection of trade-offs across tasks (ProteinGym vs PEER/DGEB, etc.).
- Education-at-scale and community resources (Academia; Education)
- Vision: Build curricula and MOOCs around CoPeP-style temporal benchmarks to train practitioners in model maintenance, data curation, and continual learning for bio.
- Potential products: Open courseware, interactive notebooks, and shared leaderboards focused on temporal data evolution.
- Assumptions/dependencies: Sustained community engagement and maintenance of datasets/tooling.
- Marketplaces for versioned biological models and update guarantees (Software/Platforms)
- Vision: Establish model marketplaces offering versioned biological foundation models with SLAs for regular updates, unlearning compliance, and task-specific variants.
- Potential products: “Biological FM Hub” with temporal badges (e.g., “UniRef2024-12 compliant,” “Safety-unlearned v3”).
- Assumptions/dependencies: Demand aggregation across organizations; interoperability standards for checkpoints and metadata.
Each application’s feasibility hinges on aligning method choice with target tasks, validating that temporal persistence is an appropriate quality proxy for the intended use, and building MLOps processes (data versioning, replay buffers, EMA resets, unlearning logs) that are auditable and sustainable at scale.
Glossary
- AdamW: An optimizer that decouples weight decay from the gradient-based update in Adam to improve generalization. "using AdamW~\citep{loshchilovDecoupledWeightDecay2018} with a weight decay of "
- AlphaFold3: A state-of-the-art, geometry-aware protein structure prediction system. "protein folding from sequence-only pLMs still lags behind specialized, geometry-aware architectures like AlphaFold3~\citep{abramsonAccurateStructurePrediction2024}."
- AMPLIFY 120M: A 120-million parameter bidirectional protein LLM used as the base model in the study. "We adopt the bi-directional AMPLIFY 120M~\citep{fournierProteinLanguageModels2024} as our base pLM for its computational efficiency."
- BERT: A bidirectional transformer architecture that inspires several protein LLMs. "draw inspiration from BERT~\citep{devlinBERTPretrainingDeep2019}."
- BiGene mining: A DGEB task category focused on mining gene–protein associations from genomic data. "including BiGene mining, evolutionary distance similarity, clustering, and retrieval."
- CASP: A biannual community challenge for assessing computational methods for protein structure prediction. "protein folding is traditionally measured on the biannual CASP challenge~\citep{jCriticalAssessmentMethods2018},"
- catastrophic forgetting: The degradation of previously learned knowledge when training on new tasks. "catastrophic forgetting, i.e., the degradation of previously acquired knowledge~\citep{mccloskey1989catastrophic, kirkpatrick2017overcoming}"
- cross-entropy loss: A standard loss function for classification or token prediction tasks that measures divergence between predicted and true distributions. "where is the cross-entropy loss"
- de novo protein design: Designing entirely new protein sequences rather than modifying existing ones. "de novo protein design~\citep{hayes2025simulating}"
- deep mutational scanning (DMS): High-throughput assays measuring the functional effects of many mutations on a protein. "217 deep mutational scanning (DMS) assays"
- DGEB (Diverse Genomic Embedding Benchmark): A multi-task benchmark spanning diverse genomic protein datasets to evaluate learned embeddings. "The Diverse Genomic Embedding Benchmark~\citep{west-robertsDiverseGenomicEmbedding2024} (DGEB) is another multi-task benchmark"
- domain-adaptive pretraining: Continual or additional pretraining on data from a specific domain to adapt a general model. "explored through domain-adaptive pretraining on distinct, specialized domains~\citep{gururanganDontStopPretraining2020, chalkidisLEGALBERTMuppetsStraight2020}"
- evolutionary distance similarity: A task evaluating similarity across sequences based on evolutionary divergence. "including BiGene mining, evolutionary distance similarity, clustering, and retrieval."
- evolutionary statistics: Statistical patterns derived from evolutionary sequence variation used to learn relationships between sequence, structure, and function. "pLMs leverage evolutionary statistics from large databases"
- exponential moving average: A smoothing technique where parameters are averaged over time with exponential decay to stabilize training. "The slow weights are an exponential moving average of the fast weights,"
- Experience Replay: A continual learning technique that rehearses samples from previous tasks to mitigate forgetting. "Standard Experience Replay~\citep{rolnickExperienceReplayContinual2019a, abbesRevisitingReplayGradient2025} mitigates catastrophic forgetting by rehearsing a small, fixed-size subset of data from prior tasks alongside the current one."
- Gradient Ascent: An unlearning method that increases the loss on a forget set to erase its influence while learning new data. "Gradient Ascent~\citep{golatkarEternalSunshineSpotless2020} takes a dual-objective approach to unlearning:"
- Hare and Tortoise: A plasticity-preserving method maintaining fast and slow weight copies, with the slow copy as an EMA of the fast. "Hare and Tortoise~\citep{leeSlowSteadyWins2024} maintains two parallel sets of network weights: fast and slow."
- i.i.d. training: Training under the assumption that samples are independently and identically distributed. "beyond that of standard i.i.d. training on individual years."
- indicator function: A function that is 1 if a condition is met and 0 otherwise, used to count membership. "where $\mathbb{1}_{D{i}(x)$ is the indicator function."
- MMseqs2: A fast and sensitive sequence search and clustering tool widely used for protein databases. "using MMSeqs2 at a sequence identity of 90\%."
- multiplicity: The number of times a sequence persists across releases, used as a proxy for reliability. "the multiplicity of a sample, with $c(x)=\sum_{i=1}^k \mathbb{I}_{D{i}(x)$."
- non-redundant clustering: Grouping sequences to remove redundancy so only representative sequences are kept. "a non-redundant clustering of UniProtKB"
- PEER (Protein Sequence Understanding): A multi-task benchmark evaluating diverse aspects of protein understanding (function, localization, structure, interactions). "The benchmark for Protein Sequence Understanding~\citep{xuPEERComprehensiveMultiTask2022} (PEER) is a multi-task benchmark"
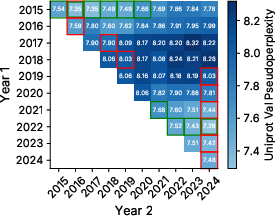
- perplexity: A language-modeling metric indicating how well a model predicts sequences; lower is better. "improves perplexity by up to 7\% even when compared to training on data from all tasks jointly."
- pLMs (protein LLMs): Models that treat amino acid sequences as language to learn protein properties. "Protein LLMs (pLMs) have recently gained significant attention"
- ProteinGym: A benchmark evaluating zero-shot prediction of mutation effects across many proteins using DMS data. "ProteinGym~\citep{notin2023proteingym} is a comprehensive fitness prediction benchmark"
- ProteinNet: A dataset framework used for protein structure prediction tasks; here referenced for contact prediction. "the ProteinNet-based contact prediction task"
- protein-ligand interactions: Interactions between proteins and small molecules or ligands, relevant for function and drug binding. "protein-protein and protein-ligand interactions."
- protein-protein interactions: Physical or functional interactions between proteins within biological systems. "protein-protein and protein-ligand interactions."
- pseudogenes: Genomic sequences resembling genes but nonfunctional, often culled during curation. "(e.g., pseudogenes)."
- Random Labeling: An unlearning technique that corrupts targets in the forget set with random labels to erase learned correlations. "Random Labeling~\citep{golatkarEternalSunshineSpotless2020} achieves unlearning by corrupting the modelâs knowledge of the forget set."
- Reference Clusters (UniRef): Periodic non-redundant snapshots of UniProt clustered at specified identity thresholds. "releases Reference Clusters~\citep{suzekUniRefClustersComprehensive2015} (UniRef),"
- sequence identity: The fraction of positions where two sequences share the same residue, often used to assess similarity. "compute the sequence identity"
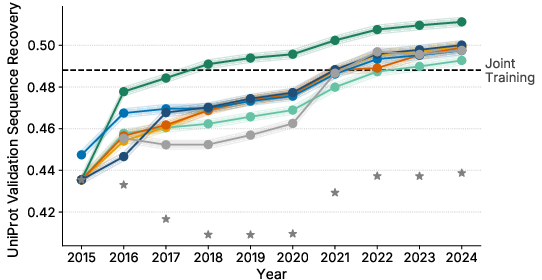
- sequence recovery: Top-1 accuracy of predicting masked amino acids in a sequence. "Sequence recovery is defined as the top-1 accuracy of masked token predictions,"
- Shrink and Perturb: A method to preserve plasticity by shrinking weights and adding noise between tasks. "Shrink and Perturb~\citep{ashWarmStartingNeuralNetwork2020} mitigates the loss of plasticity by periodically scaling down network weights and injecting noise."
- Spearman's rank correlation (ρ): A nonparametric rank correlation metric used to evaluate predicted fitness rankings. "using Spearman's rank correlation ()."
- subcellular localization: The specific location within a cell where a protein resides, influencing its function. "function, subcellular localization, structural properties,"
- temporal importance sampling: Sampling strategy that biases replay towards examples persisting across time. "we modify this approach to use an unbounded replay buffer with a temporal importance sampling strategy."
- temporal meta-information: Information derived from the history of database entries (e.g., persistence over releases). "incorporating temporal meta-information improves perplexity by up to 7\%"
- Temporal Replay: A replay strategy that prioritizes historically persistent sequences during continual pretraining. "Temporal Replay achieves the best performance"
- UniProt Knowledgebase (UniProtKB): A comprehensive database of protein sequence and functional information, continuously curated and updated. "the UniProt Knowledgebase~\citep{theuniprotconsortiumUniProtUniversalProtein2025} (UniProtKB),"
- UniRef100: UniProt Reference Clusters with 100% identity threshold where each cluster is represented by its longest sequence. "The CoPeP benchmark is composed of 10 consecutive UniRef100 yearly releases from 2015 to 2024"
- unlearning: Techniques to remove the influence of specific data from a model after training. "Unlearning aims to actively erase knowledge about specific samples from the network."
- Warmup-Stable-Decay (WSD): A learning rate schedule with warmup, a long plateau, and a final decay phase. "replace the cosine decay with Warmup-Stable-Decay~\citep{huMiniCPMUnveilingPotential2024, liModelMergingPretraining2025} (WSD)."
- wild-type sequence: The most common or reference sequence of a protein used as a baseline for mutational studies. "For each wild-type sequence"
- zero-shot setting: Evaluating a model on tasks or distributions without task-specific training or fine-tuning. "predicted in a zero-shot setting."
Collections
Sign up for free to add this paper to one or more collections.