- The paper introduces a framework where path integrals map quantum systems onto classical ring polymers to accurately capture nuclear quantum effects.
- It details acceleration techniques like ring polymer contraction and higher-order Trotter splittings to address computational challenges.
- The approach offers practical guidelines for leveraging path integral molecular dynamics to simulate thermodynamic and dynamical properties of complex systems.
Detailed Technical Essay: Path Integral Methods in Atomistic Modelling
Introduction
The paper "Path Integral Methods in Atomistic Modelling: An Introduction" (2603.28588) serves as a comprehensive pedagogical exposition of path integral (PI) techniques for modelling quantum nuclear effects (NQEs) in atomistic simulations. The focus is on the theoretical underpinnings, computational techniques, and practical considerations associated with Feynman path integrals, their classical isomorphism, and applications to thermodynamic and dynamical properties—providing both conceptual clarity and methodological pragmatism for the computational physics and chemistry communities.
Classical and Quantum Statistical Mechanics Foundations
The authors begin by rigorously recasting the statistical mechanics of N-body systems. In the canonical ensemble, classical statistical sampling is performed over a phase space (p,q) with the Boltzmann measure, where the configurational part is typically intractable for high-dimensional systems. This motivates Markov Chain Monte Carlo (MCMC) methods and molecular dynamics (MD) as canonical sampling engines. MD obeys Hamiltonian dynamics, preserving both phase space volume and the correct distributional invariants, a property exploited in modern extended-ensemble and thermostatted schemes.
The Langevin equation, introduced as a stochastic extension to canonical sampling, incorporates random and frictional forces representing environmental coupling. Its stochastic calculus formulation, Fokker–Planck description, and autocorrelation analysis (including the ergodicity–sampling error relationship) are presented, culminating in practical guidance for the use of thermostats (e.g., BAOAB) and autocorrelation-based trajectory analysis.
Feynman Path Integrals and Classical Isomorphism
Central to the PI formalism is the imaginary-time path integral representation of the quantum partition function and propagators. The paper introduces the Trotter product decomposition, leading to a discretized imaginary-time path where the quantum system maps onto a classical ring polymer—a configuration of P classical replicas (beads) connected by harmonic springs, each bead corresponding to a discretized time slice in imaginary time.
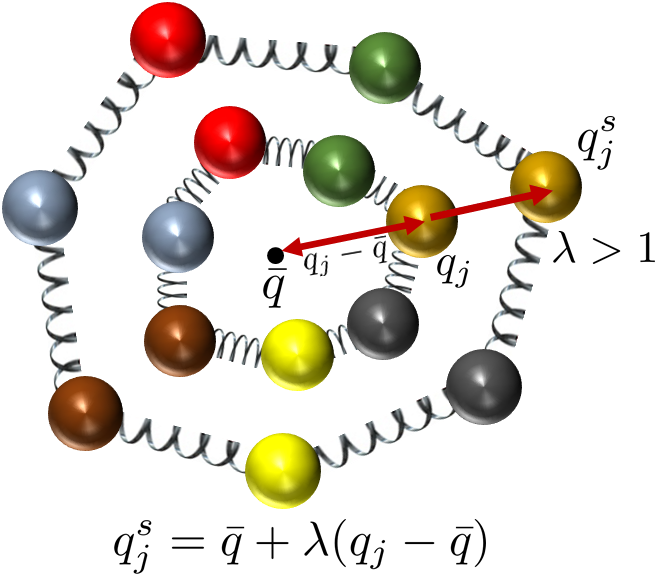
Figure 1: Schematic of a coordinate-scaled ring polymer configuration, which forms the basis for virial estimators in PIMD.
This mapping (the classical isomorphism) forms the basis of path integral molecular dynamics (PIMD). The quantum partition function thus becomes a high-dimensional classical integral over replicated degrees of freedom, enabling the use of MD and MCMC sampling for quantum systems. The convergence of this mapping is controlled by the number of replicas P, which must resolve all relevant quantum (vibrational) frequencies. The computation cost scales linearly with P; for stiff systems (e.g., high-frequency OH stretches in water), P can be large, motivating the development of acceleration schemes.
A key technical challenge is estimation of observables, particularly those involving the kinetic energy operator. The primitive estimator, derived via thermodynamic differentiation, suffers from growing variance with increasing P due to statistical cancellation between large terms. The paper describes the virial estimator, derived via coordinate scaling of the partition function, which circumvents this problem by exploiting ring-polymer centroid properties.
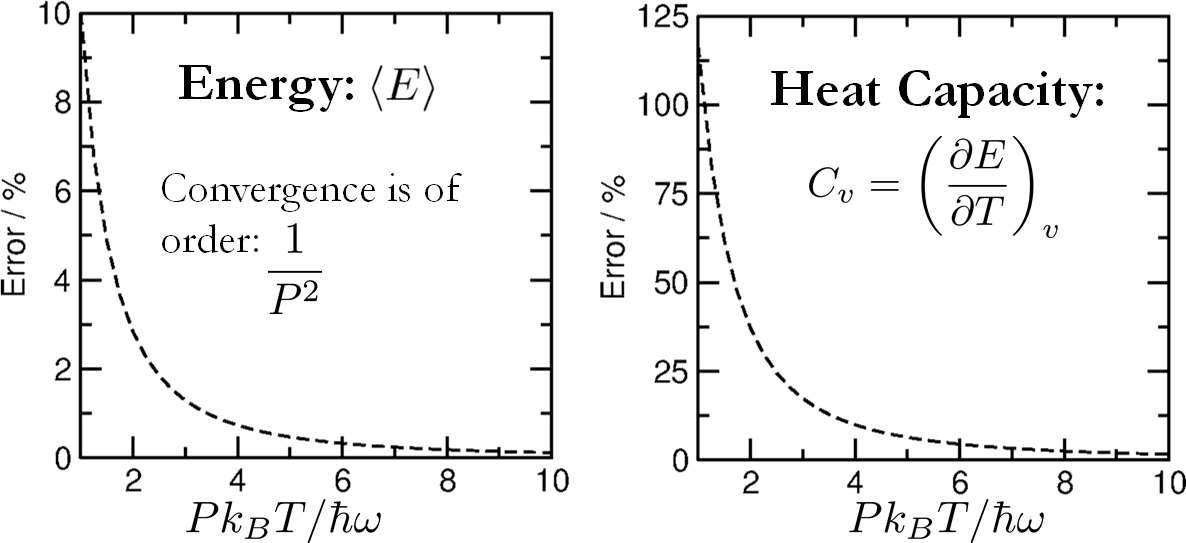
Figure 2: Percentage error in energy and heat capacity for a harmonic oscillator as function of PkBT/ℏω; heat capacity convergence requires more beads due to second-moment sensitivity.
For momentum-dependent observables or off-diagonal elements, the path integral formalism must treat open-chain ring polymers, as depicted below:
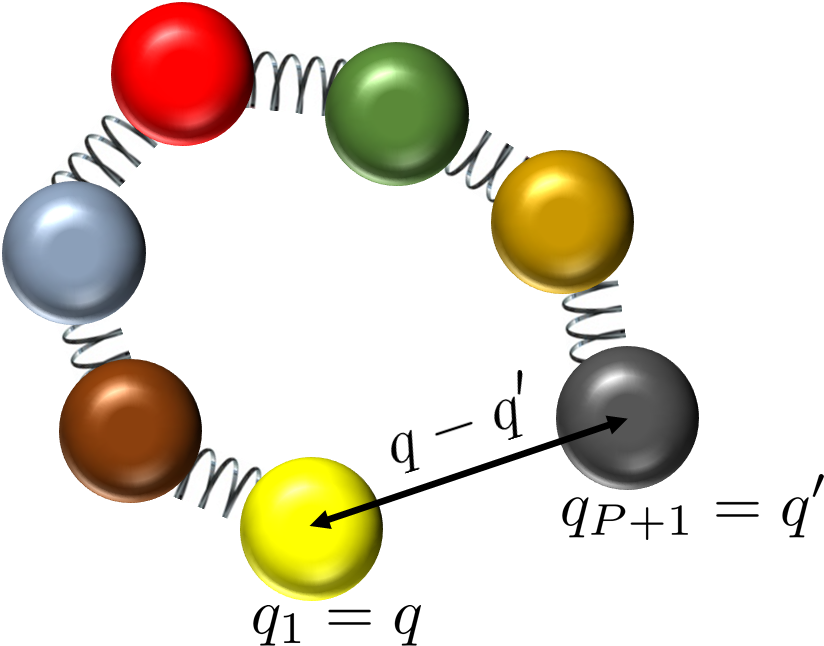
Figure 3: Open-chain ring polymer configuration with end-to-end distance relevant for momentum distribution estimators.
Fourier transforms of the end-to-end distance yield quantum momentum distributions, key for interpreting deep inelastic neutron scattering data.
Convergence Control and Acceleration Techniques
Efficiency of PI methods in practice is governed by the number of beads needed to converge various observables, which directly relates to the highest vibrational frequency ωmax present in the system:
P>kBTℏωmax
For liquid water at 300 K, high-frequency OH stretches drive P>20 (as shown in Figure 4), while low-frequency modes require far fewer replicas.
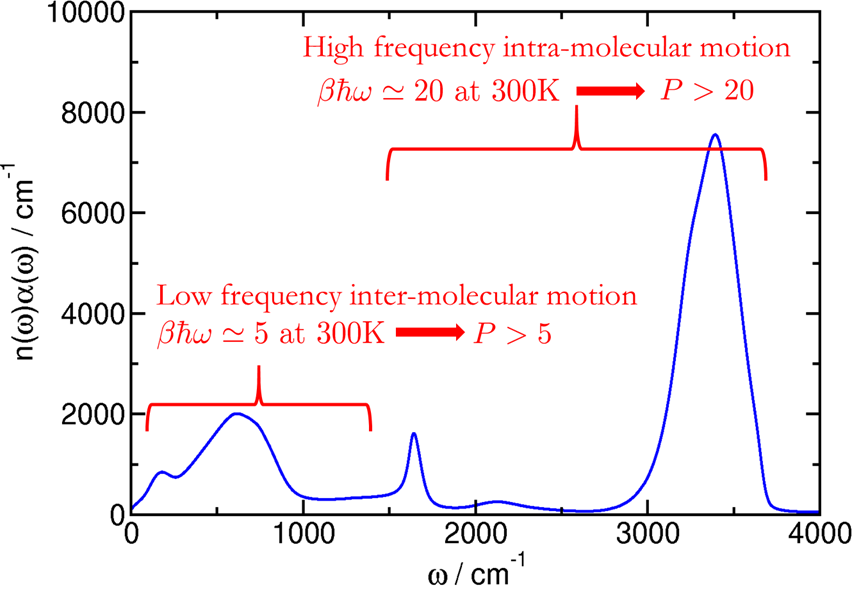
Figure 4: IR absorption spectrum of liquid water showing that low-frequency intermolecular modes can be converged with P≳5, but high-frequency stretches require P>20.
Ring polymer contraction (RPC) and related multilevel schemes exploit the frequency dependence of P, evaluating stiff components of the potential on a coarser (contracted) representation and projecting forces back onto the full polymer, as systematically outlined in:
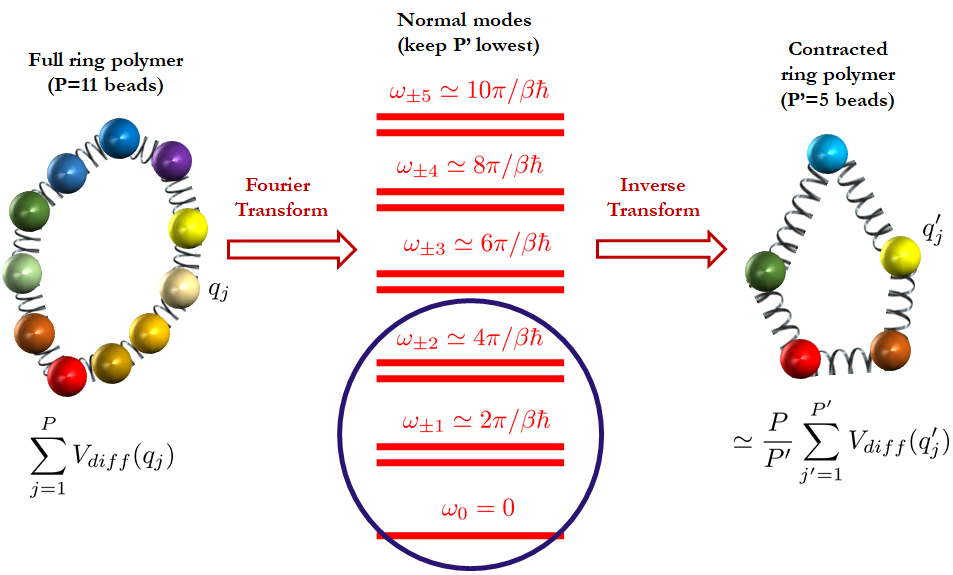
Figure 5: Outline of the RPC procedure, showing contraction and expansion between full and reduced normal mode representations.
Higher-order Trotter–Suzuki splittings (Takahashi-Imada, Suzuki-Chin, etc) further accelerate convergence, reducing global error from O(P−2) to O(P−4), albeit at the cost of requiring force and even Hessian information for the system, posing challenges in ab initio simulations.
Quantum Dynamics Approximations: RPMD and Its Limitations
The RPMD (ring polymer molecular dynamics) approach is presented as a practical route to quantum real-time dynamics within the PI framework. It is formally exact at short times and for correlation functions of linear operators in harmonic systems, provides the exact quantum rates for parabolic barriers above the crossover temperature, and yields accurate diffusion coefficients and orientational relaxation times for liquids.
However, the approach is fundamentally limited for high-frequency vibrational spectra and nonlinear operator correlation functions due to contamination from internal polymer modes, resulting in "spurious resonances." Advanced thermostatted methods (TRPMD, CMD) and deconvolution procedures partially mitigate these deficiencies in the computation of vibrational spectra.
Generalized Langevin Equations (GLEs) and Non-Equilibrium Methods
The introduction of colored-noise thermostats via GLEs allows for frequency-dependent thermostatting, crucial for efficient PIMD, stabilizing multiple time-stepping (MTS) algorithms, and even for modelling non-equilibrium systems (e.g., δ-thermostat for selective mode excitation).
GLEs can be constructed to enforce quantum mechanical fluctuations mode-wise ("quantum thermostat"). The coupling of GLE to PIMD (PI+GLE, PIGLET) accelerates convergence to the quantum limit by matching bead correlation structure not just marginally, but in higher-order cumulants required for correctly sampling the virial estimator and other bead-bead sensitive observables.
Implications, Prospects, and Future Directions
Path integral methods, as systematically laid out in this work, provide a rigorous and extensible framework for including nuclear quantum effects in atomistic simulations. The methodologies enable the quantitative prediction of isotope effects, zero-point vibrational energies, tunneling rates, and quantum-corrected structural and thermodynamic properties in molecular and condensed systems.
The paper positions PIMD, PIGLET, and RPMD as core tools for the simulation of molecular materials, while highlighting the ongoing need for robust acceleration (RPC, high-order Splittings, GLEs), continued improvements in quantum dynamics approximations, and sophisticated estimator construction for complex observables such as isotope fractionation, vibrational spectra, and nonequilibrium response.
Integration with machine-learned potentials and scalable force fields will further enhance the applicability of these frameworks, enabling fully quantum simulations of large heterogeneous systems. The modularity and theoretical robustness of the PI approach guarantees its centrality in the next generation quantum molecular mechanics and material modelling toolkits.
Conclusion
This work provides a methodologically rigorous yet accessible resource for advanced researchers undertaking quantum statistical mechanics simulations. By bridging theoretical derivation and practical implementation across a range of estimators, sampling protocols, and acceleration techniques, it establishes a foundation for continued advances in the simulation of complex quantum materials and molecular systems. The adoption and continued refinement of PI-based methods will be a driver for future breakthroughs in chemical physics, materials engineering, and quantum simulation.