- The paper introduces the evo-SIS model that integrates stochastic transmission, mutational diffusion, and immune-mediated competition to explain pathogen diversity.
- It demonstrates through simulations that epidemic size and strain evenness sharply increase as transmission surpasses a critical threshold.
- Empirical validations with COVID-19 data confirm the model’s predictions and its utility in guiding vaccination strategies and outbreak preparedness.
Coevolutionary Dynamics and Emergence of Pathogen Diversity in Interconnected Systems
Introduction
"Pathogen diversity emerging from coevolutionary dynamics in interconnected systems" (2603.29398) formulates a multiscale theoretical framework capturing the interplay between pathogen evolution and epidemic dynamics across heterogeneous metapopulation networks. The approach blends stochastic transmission, mutational dynamics, and immune-mediated competition using networks to model pathogen strains and host populations. Central to the evo-SIS model are two intertwined structures: a metapopulation network encoding spatial or social heterogeneity and a mutation network representing the strain space, with cross-immunity rendered as diffusion distances.
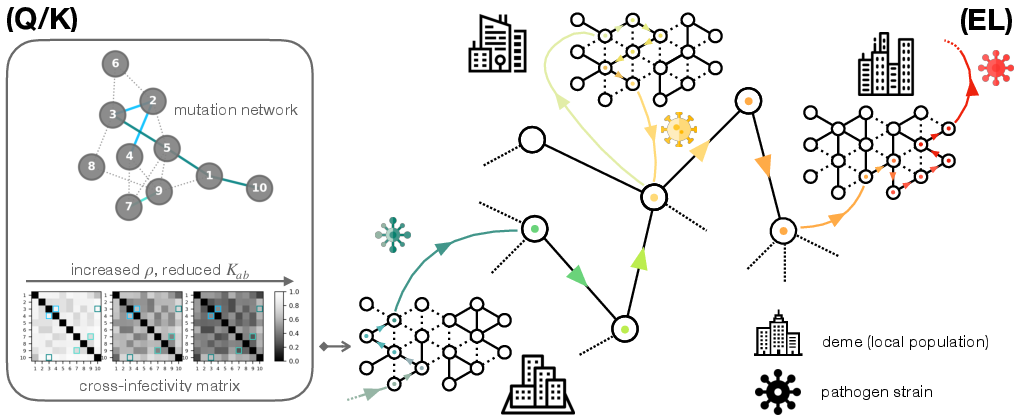
Figure 1: The evo-SIS framework depicts mutations as diffusion on a strain network (Q/K) and their interplay with pathogen spread across a metapopulation (EL) giving rise to emergent evolutionary landscapes.
The evo-SIS model considers N demes (subpopulations) interconnected via an adjacency matrix A and M pathogen strains with mutational connectivity Q. Infection dynamics occur at strain-independent rates, but are modulated by a cross-infectivity matrix K, calculated as the diffusion distance in the mutation network. Mutation events are modeled as diffusion regulated by the Laplacian LQ.
The core differential equations for infected (Ixa) and susceptible (Sxa) fractions per deme and strain encode infection, recovery, and mutation, with cross-immunity embedded via K:
I˙xa=βy,b∑AxyKbaSxbIya−Ixa−ηb∑(LQ)abIxb
A0
This structure permits generalization to arbitrary mutation networks (undirected or directed), multi-parameter cross-immunity, and metapopulation heterogeneity.
Critical Phenomenology and Pathogen Diversity
The model admits three principal regimes: extinction, recurrent outbreaks, and endemic persistence, controlled by the effective transmission parameter A1 relative to a critical threshold A2. The strain turnover dynamics and epidemic size (total prevalence) are augmented by the mutation rate A3 and cross-immunity parameter A4, which adjusts the topology and geometry of A5.
Evenness, defined as normalized entropy of strain prevalences, serves as an effective diversity metric:
A6
Simulation results demonstrate that as A7 crosses A8, both epidemic size and evenness sharply increase, with high mutation and moderate A9 producing sustained strain diversity. Notably, the model reveals a non-monotonic dependence of prevalence on heterogeneity, with maximal epidemic size at intermediate levels.
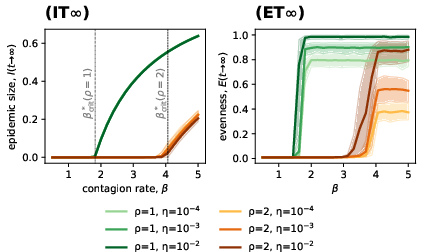
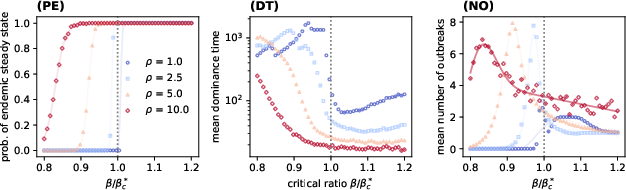
Figure 2: Asymptotic epidemic size and evenness versus M0, showing sharp transitions near the critical threshold with quantification across strain ensemble realizations.
Empirical Validation and Mutational Structure Effects
By comparing model outputs to empirical COVID-19 datasets—specifically strain prevalence time series across multiple countries—the evo-SIS model replicates observed patterns of consecutive predominant strains, outbreak sizes matching empirical peaks, and dominance times consistent with data. When the mutation network is directed (enforcing temporal exclusion of earlier strains via triangular matrices), the model more closely mirrors real-world exhaustion of early strains and lack of back-mutations.
Figure 3: Model predictions contrasted with COVID-19 strain time series across six countries, highlighting both undirected and directed mutation structures and dominance trajectories.
Steady-State Analysis and Evolutionary Landscape
Near endemic steady states and under large force of infection, the dynamics reduce via adiabatic elimination to a replicator-mutator-like form governing strain composition:
M1
where the fitness component M2 is a nonlinear function of instantaneous strain composition and cross-infectivity structure.
Higher-order corrections link epidemic size directly to strain composition changes, and reveal multiplicative antigenic competition effects, handling both selection and mutation drift. Simulations confirm that leading-order and next-order approximations adequately capture steady-state diversity and transient epidemic variations.
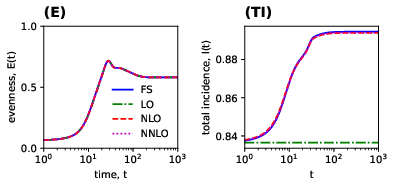
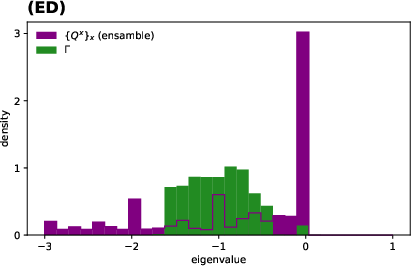
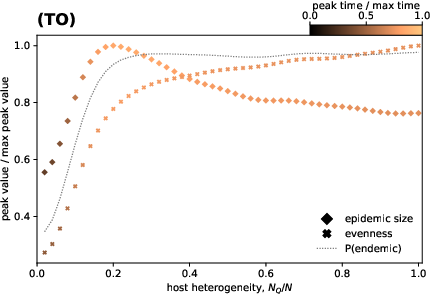
Figure 4: Approximant dynamics near endemic equilibrium capture evenness and variation in total incidence; spectral analysis at the metapopulation level reveals emergence of a dynamical evolutionary landscape.
Heterogeneous Host Populations and Extended Evolutionary Paths
Extending the framework to heterogeneous demes, each with distinct mutation matrices (M3), the coupling across demes facilitates exploration of otherwise disconnected strain space components. Numerical eigenvalue density analyses of the evolutionary landscape Laplacian validate the emergence of new connectivity, producing a spectral gap absent from isolated mutation matrices. Strain diversity increases monotonically with heterogeneity, while prevalence exhibits a peak at intermediate diversity ratios.
Figure 4: Host heterogeneity induces monotonic increases in evenness and non-monotonic epidemic size; spectral gap in evolutionary landscape highlights emergence of interconnected strain space.
Within-Host Dynamics and Mutation Network Directionality
Appendix analyses leverage replicator-mutator dynamics at the within-host level, where fitness is suppressed by immune memory, generating deterministic cycles of dominant strain successions. Directed mutation networks emerge naturally from multiple immune memories, enforcing temporal ordering and blocking re-emergence of earlier strains—critical for realistic modeling of pathogens such as influenza and SARS-CoV-2.
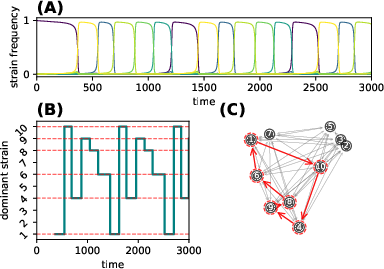
Figure 5: Deterministic cycles of strain dominance in within-host replicator-mutator dynamics demonstrate the emergent directed structure of mutation networks under immune-mediated selection.
Diffusion Geometry and Cross-Immunity Quantification
The model's operationalization of cross-immunity is grounded in diffusion geometry, with diffusion distance between strains encoding antigenic similarity. The degree-weighted Laplacian is preferred for phenotypic similarity, while the unweighted Laplacian governs mutational drift, reflecting assumptions regarding holding times and transition probabilities.
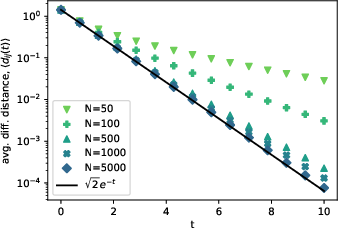
Figure 6: Diffusion distance behavior over time for Erdos-Renyi networks reveals the analytical structure underpinning cross-immunity and epidemic thresholds.
Implications and Future Directions
The evo-SIS framework provides a minimal yet flexible structure to disentangle the coevolutionary feedbacks between mutation, transmission, and immune competition. It isolates the effects of network topology, host and strain heterogeneity, and the emergence of directional mutational landscapes resulting from immune memory. Practically, the model informs strategy design for vaccination and outbreak preparedness, highlighting the impact of metapopulation connectivity and host diversity on the potential for antigenic escape.
Theoretical extensions are warranted to integrate mechanistic within-host diversity, lineage divergence, co-infection, and explicit mapping between mutational structure and antigenic similarity. Further exploration of adaptive landscapes and their eigenstructure, alongside refined empirical calibration, will deepen insights into pathogen evolution and epidemic control. The framework is directly relevant to AI-aided epidemiological modeling and synthetic biology, where network-based approaches are increasingly adopted for prediction, intervention, and risk management.
Conclusion
This study rigorously couples multistrain epidemic dynamics with network-based mutation and spatial heterogeneity, offering a comprehensive, scalable foundation for understanding pathogen diversity. By merging leading principles from replicator-mutator theory, diffusion geometry, and network epidemiology, it elucidates how immune-mediated competition and metapopulation structure drive both the transient and long-term diversification of pathogens. The model's minimalism, adaptability, and explicit connections to empirical phenomena render it a valuable tool for ongoing and future pathogen surveillance, evolutionary forecasting, and behavioral modeling initiatives.