- The paper demonstrates that DRISM with pair-specific LJ parameters greatly enhances the accuracy of modeling EDL structure and CO adsorption energetics.
- It employs quantum-chemical simulations to benchmark DRISM against Poisson–Boltzmann and explicit MD, producing detailed solvent and ionic density profiles.
- Findings highlight that parameter sensitivity critically impacts differential capacitance and solvation predictions, underscoring the need for refined implicit modeling.
Quantum-Chemical Modeling of the Electrochemical Double Layer via DRISM and Implicit Solvation
Introduction
Accurate atomistic modeling of the electrochemical double layer (EDL) is critical for understanding and quantitatively predicting interface-dependent phenomena such as electrocatalysis, energy storage, and charge transfer at electrode-electrolyte interfaces. While first-principles methods such as ab initio molecular dynamics (AIMD) provide highly detailed descriptions, their unfavorable scaling, limited sampling, and slow convergence at constant-potential pose major challenges for routinely studying realistic electrified interfaces. Conversely, implicit and continuum solvation approaches—most notably the Poisson–Boltzmann (PB) framework—offer computational efficiency but neglect essential molecular granularity and correlation effects.
This work systematically investigates and benchmarks the dielectrically-consistent reference interaction site model (DRISM) as an advanced implicit electrolyte formalism for EDL modeling in quantum-chemical simulations. DRISM serves as a rigorous statistical mechanical alternative to PB for embedding quantum systems, enabling direct computation of solvent and ionic density profiles. The study focuses on a paradigmatic gold-electrolyte interface with aqueous $\ce{NaCl}$, comparing DRISM to PB and explicit molecular dynamics (MD) references across density profile, differential capacitance, and adsorption energetics regimes. The influence of Lennard-Jones (LJ) parametrization and the application or modification of Lorentz–Berthelot (LB) mixing rules versus pair-specific interactions is assessed in detail.
Solvent and Ionic Density Profiles
Quantum-mechanical (QM) DRISM calculations yield solvent and ionic distributions as ensemble-averaged, x-y slab density profiles as functions of separation from the electrode. For the $\ce{Au}(111)$ facet, DRISM-based water profiles show reasonable spatial agreement with explicit MD (GolP-CHARMM force field, mTIP3P water) in the location of major hydrogen and oxygen peaks, though DRISM underestimates peak magnitudes, consistent with known KH closure artifacts.
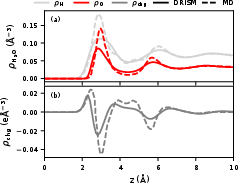
Figure 1: Hydrogen (light gray) and oxygen (red) particle density profiles for $\ce{Au}(111)$/water; DRISM (solid) and MD (dashed) with mTIP3P.
A direct comparison to PB modeling highlights the molecular oscillations captured by DRISM versus the smeared, single-peak bound charge structure associated with continuum dielectrics.
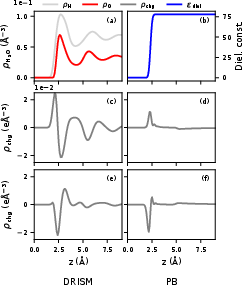
Figure 2: Solvent modeling in PB versus DRISM; DRISM captures molecular-layer oscillations absent in PB.
For the ionic profiles, the DRISM framework exposes a pronounced sensitivity to choice of LJ parameters, in particular for the metal-ion interaction. For standard LJ mixing, notably LB, DRISM predicts significant $\ce{Na+}$ accumulation directly at the inner Helmholtz plane (IHP), especially at negative electrode charge. This trend is exaggerated with strong gold–ion LJ well depth parameters (Heinz et al.) and is suppressed for weaker (e.g., UFF) parametrizations.
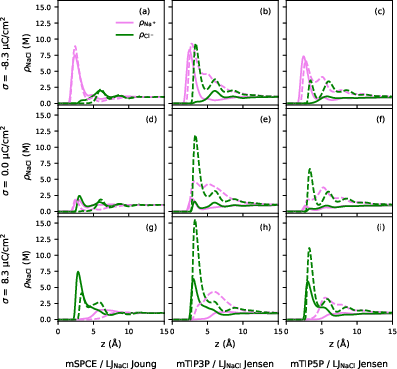
Figure 3: Ionic density profiles for $\ce{Au}(111)$/1 M $\ce{NaCl}$ under various parametrizations and surface charges.
Cross-validation with explicit MD simulations confirms the qualitative correctness in the position of both cation and anion peaks, though DRISM’s default LB mixing rule can yield unphysically high interfacial cation densities.
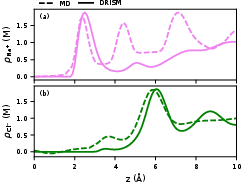
Figure 4: Comparison of DRISM and explicit MD density profiles for $\ce{Au}(111)$/1 M $\ce{NaCl}$ at the PZC; major peak positions agree.
When pair-specific metal–ion LJ parameters are incorporated, as opposed to standard LB combination rules, DRISM can recover the expected exclusion (Stern-type) layer at the interface and eliminate the anomalous cation peak.
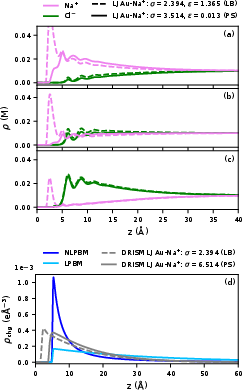
Figure 5: Density profiles for x0/0.01 M electrolyte; LB rules (dashed) yield excess cation at the IHP, while pair-specific parameters (solid) restore exclusion.
At large distances from the interface, DRISM correctly displays an exponential decay of ionic charge density into the diffuse layer, quantitatively reproducing the crossover between linear and nonlinear PB regimes.
Differential Capacitance
The study reports the computed differential capacitance (DC) as a primary observable connecting interfacial structure to thermodynamic response, mapping DC versus electrode potential for x1/x2 and x3 at various concentrations and parameter sets.
DRISM with LB mixing and common x4 parametrizations predicts a steep, unphysical increase (divergence) in DC at negative potentials (anodic branch) for concentrated electrolytes (x51 M), resulting from spurious cation condensation at the interface.
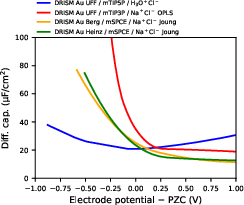
Figure 6: Differential capacitance for x6/1 M aqueous electrolyte; excessive DC peak at negative potentials due to unphysical x7 accumulation.
This divergence is not present with x8 as cation, nor is it supported by experiment for comparable systems. The DC is rapidly suppressed upon reducing electrolyte concentration, with negligible anomalies for x9 M, and is further regularized by introduction of pair-specific gold–cation LJ parameters, which shift the excess cation peak away from the interface.
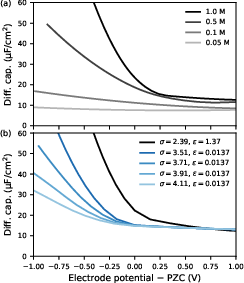
Figure 7: Differential capacitance for y0/aqueous y1: (a) concentration dependence; (b) effect of pair-specific y2–y3 parameters—strong suppression of unphysical DC peak.
Importantly, DRISM does not reproduce experimentally observed cation-specific trends in DC magnitude at the PZC for different alkali metals, in contrast to modern measurements on noble metal electrodes. DRISM-predicted DC is essentially cation-independent within the parameter space explored, a marked limitation in capturing cation hydration effects.
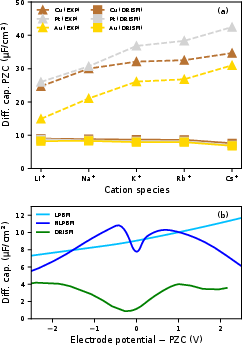
Figure 8: (a) DC at PZC for (111) facets of y4, y5, and y6 for various cations (DRISM: squares; experiment: triangles); (b) full DC curve vs. PB at 0.01 M.
At dilute concentrations, the overall DC profile is in qualitative but not quantitative agreement with PB. The minimum in DC is shifted and the magnitude is generally underestimated relative to PB, potentially due to reduced Helmholtz layer contribution in DRISM.
Solvation Effects in CO Adsorption
The paper examines the influence of solvation and electrolyte parameters on the computed adsorption energies of CO on y7 surfaces. DRISM predicts a strong and systematic dependence of solvation stabilization/destabilization on the chosen metal–water LJ parametrization, with differences up to y8 kcal/mol across parameter sets.
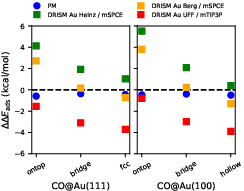
Figure 9: Solvation contribution to the adsorption energy of CO at y9 and $\ce{Au}(111)$0 as function of parametrization; Poisson yields negligible stabilization, while DRISM response is substantial and parameter-dependent.
DRISM with Heinz parameters destabilizes adsorption significantly, while UFF parameters provide stabilization. This behavior is rationalized by the differences in $\ce{Au}(111)$1–$\ce{Au}(111)$2 association energies in the respective LJ schemes. Trends across CO adsorption sites are consistent, but magnitude is highly sensitive to parametrization. The inclusion of a mobile electrolyte ($\ce{Au}(111)$3 M $\ce{Au}(111)$4) has a clear impact: spurious asymmetry and enhanced (unphysical) solvation penalty for CO adsorption at negative potential when default LB rules are used, mirroring the corresponding DC anomalies. Application of pair-specific gold–ion parameters restores the expected symmetry around the PZC in the potential-dependent CO adsorption free energy.
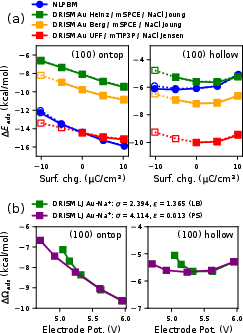
Figure 10: (a) CO adsorption energy at $\ce{Au}(111)$5/1 M electrolyte as a function of surface charge; (b) grand-canonical adsorption energy vs. potential with LB (green) and pair-specific (purple) parameters. Pair-specific tuning restores symmetry around the PZC.
Implications and Future Prospects
The results demonstrate that QM/DRISM, when combined with careful, pair-specific parametrization of metal–water and metal–ion LJ potentials, can semi-quantitatively reproduce critical aspects of EDL structure, DC, and solvation energetics. However, reliance on default or empirically inherited mixing rules leads to qualitatively incorrect predictions—particularly excessive cation condensation at negative potentials and corresponding artifacts in DC and adsorption energetics.
The inability of DRISM in its current implementation to recover experimentally observed cation series trends in DC, as well as its overdependence on parametrization rather than ab initio derived interfacial physics, underscores an outstanding limitation relative to both explicit MD and advanced mean-field approaches. Extension of the DRISM formalism to incorporate polarization effects, explicitly many-body metal–ion potentials, and possibly data-driven parameter optimization is warranted. Future developments should target a more systematic and transferable parametrization protocol for QAIMD embedding or ab initio force field mapping—potentially leveraging modern machine learning force field technologies.
The comparative analysis with Poisson–Boltzmann and explicit MD indicates that, for both theoretical and practical electrochemical modeling, the best utility of DRISM will arise when used as a flexible, controlled intermediary between continuum models and explicit sampling approaches.
Conclusion
This study establishes the DRISM embedding formalism as a powerful and efficient framework for implicit electrolyte modeling in quantum-chemical EDL simulations. The sensitivity of DRISM to the details of the metal–ion and metal–solvent interaction parameterization is pronounced; default mixing approaches (LB) are generally inadequate, particularly for strongly interacting or multicomponent systems. Use of pair-specific parameters is shown to be essential for the physically accurate description of the compact EDL structure, suppression of anomalous cation accumulation at the IHP, and correction of artifactually large or asymmetric differential capacitance and solvation energies.
DRISM robustly interpolates between molecular granularity and mean-field PB diffuse layer predictions and, with refined parametrization, is positioned as an advanced tool for investigating composition- and potential-dependent properties at electrified interfaces. However, full fidelity to experimental trends and explicit simulation remains contingent on the further development of parametrization strategies, possibly involving ab initio and data-driven force field generation.
Key conclusion: Adoption of pair-specific LJ parameters in QM/DRISM greatly enhances the physically realistic modeling of EDL structure, differential capacitance, and solvation contributions, opening avenues for systematic parameter optimization and improved implicit solvent modeling for complex electrochemical systems (2603.29674).