- The paper introduces an ML-driven framework that optimizes chemical discovery through efficient data usage and active learning strategies.
- It demonstrates that equivariant force fields and foundation models significantly reduce training data requirements while improving transferability across chemical systems.
- The study outlines automated, self-driving research pipelines that integrate physical constraints and active sampling to minimize environmental costs.
Sustainable Exploration of Chemical Spaces with Machine Learning: Efficiency, Data, and Automation
Introduction and Sustainability Landscape
The integration of artificial intelligence into molecular and materials science has led to a paradigm shift in discovery, analysis, and automation. However, the exponential increase in computational and data requirements introduces environmental and societal sustainability challenges, including rising energy consumption and carbon emissions. This work systematically evaluates the end-to-end sustainability aspects of ML-driven chemical discovery, focusing on the lifecycle from quantum mechanical (QM) data generation to automated, self-driving research workflows.
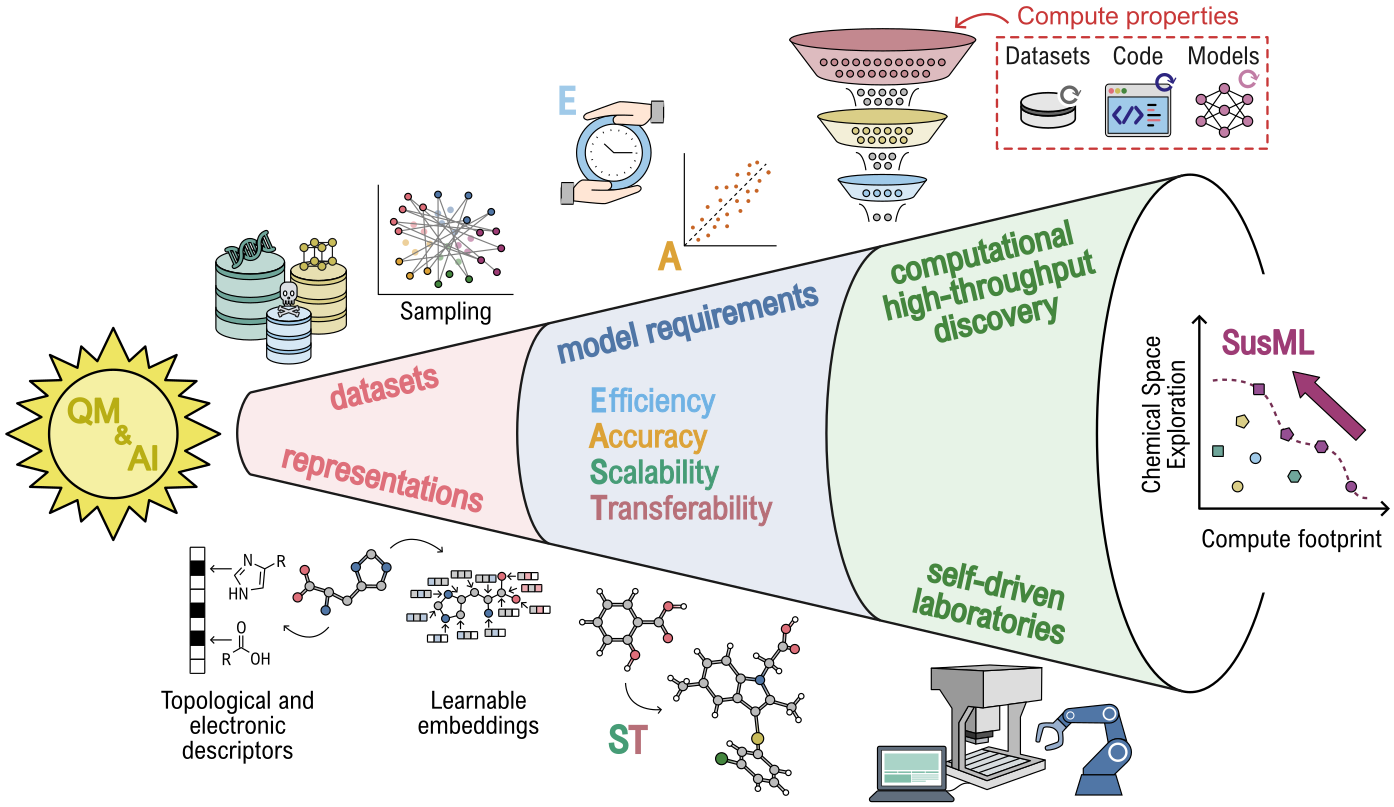
Figure 1: Scheme illustrating the sustainability topics discussed at the SusML workshop (Dresden, Germany), covering the AI-driven discovery pipeline from QM data generation to automated workflows.
The framework is anchored in strategies such as general-purpose ML models, multi-fidelity approaches, active learning, the incorporation of physical constraints, and open dissemination of data and tools. The focus is not only on methodological advancements but also on practical, domain-specific implementations to reduce overall computational and environmental costs.
Equivariant ML Force Fields and Foundation Models
Equivariant machine learning force fields (MLFFs) leverage symmetry-aware representations, notably through message-passing neural networks and Graph Atomic Cluster Expansion (GRACE), achieving superior data efficiency and transferability across diverse chemical systems. Compared to non-equivariant counterparts, these models consistently demonstrate higher generalizability and accurate reproduction of reference ab initio dynamics even at reduced training set sizes, largely due to their geometric priors and formal completeness [batatia2022mace, bochkarev2024graph, batatia2025design, lysogorskiy2025graph].
Extending this, foundation models trained on extensive, openly available datasets (e.g., QM9, Materials Project, OMat24, Alexandria) [Ramakrishnan2014, BarrosoLuque2024Open, cavignac_ai-driven_2025] enable direct out-of-the-box predictions and efficient fine-tuning for new chemical domains, amortizing the high upfront resource cost over a wide range of applications. Nevertheless, inference costs, especially for expressive equivariant models, remain a concern; distillation and optimized kernel engineering strategies are critical to practical large-scale deployment [Gardner2025Distillation].
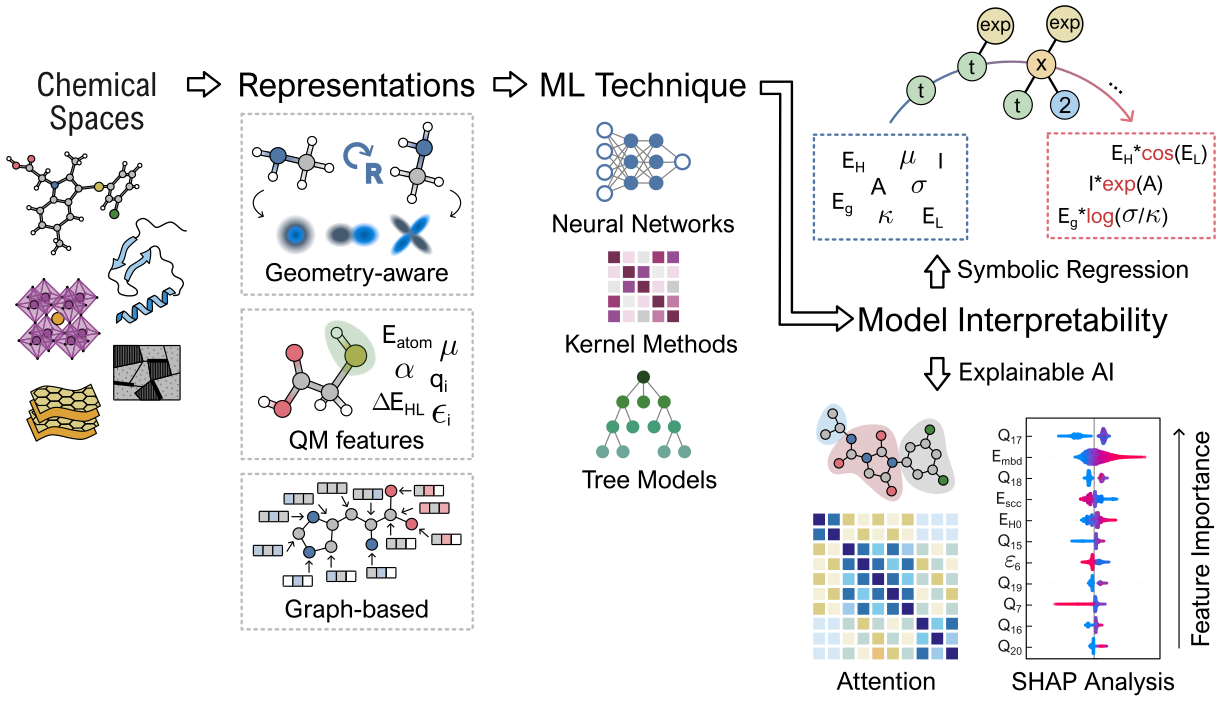
Figure 2: Workflow for predictive modeling via quantum-inspired representations, ML models, and explainability techniques.
For property prediction in drug discovery and materials informatics, enhanced representations that include quantum mechanical descriptors (e.g., orbital energies, charges) have demonstrated significant data efficiency gains and interpretability improvements in low-data and out-of-distribution regimes [hinostroza2026assessing, cremer2023equivariant, bose2026quantum]. For sparse data scenarios typical in materials informatics, parsimonious symbolic learning approaches such as SISSO afford interpretable, physically grounded structure-property relationships that require only modest dataset sizes and yield closed-form predictive models, facilitating sustainable large-scale screening and deeper insights into property origins.
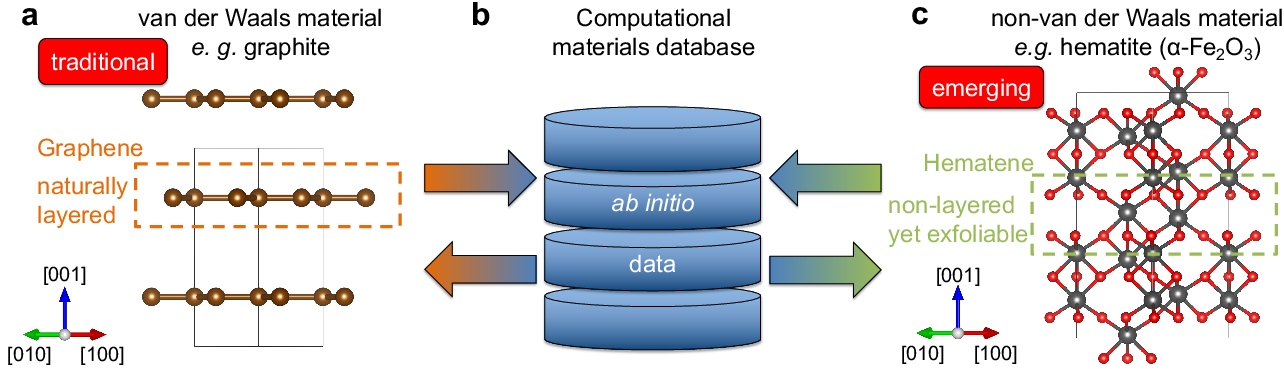
Figure 3: Traditional vs. emerging 2D materials: (a) classical vdW 2D systems, (b) effect of databases, (c) early-stage ML for non-vdW 2D materials.
The treatment of complex, strongly correlated or multiconfigurational electronic states remains challenging for ML. High-fidelity solvers and explicitly designed features that reflect spin, degeneracy, and entanglement are essential. ML surrogates are best deployed hierarchically to ensure physically meaningful predictions only in well-posed regions, supported by indicators of electronic complexity [Unke2021, King2025, Dobrautz2024].
Sustainable Expansion and Inverse Design with Generative Models
Generative AI for Molecules and Materials
Generative learning approaches—encompassing variational autoencoders, diffusion models, adversarial networks, and genetic algorithms—have moved beyond purely forward high-throughput screening, enabling the inverse (property-driven) design of molecules and materials. Such frameworks have been tailored to transition metal complex chemistry, porous materials, and solid-state systems, where experimental or computational data availability is sparse and where stringent multi-objective or physical constraints (e.g., stability, synthetic accessibility) must be incorporated [Kneiding263, Cornet1793, zhaoHighThroughputDiscoveryNovel2021, zeniGenerativeModelInorganic2025].
For crystal generation, the development of robust, invertible, and symmetry-invariant representations is a critical outstanding challenge. Novel string-based (e.g., SLICES), matrix, and symmetry-based encodings have been proposed to capture the structural richness of periodic materials efficiently, yet current models still struggle with disorder, defects, and explicit synthetic considerations [wangLeveragingGenerativeModels2025].
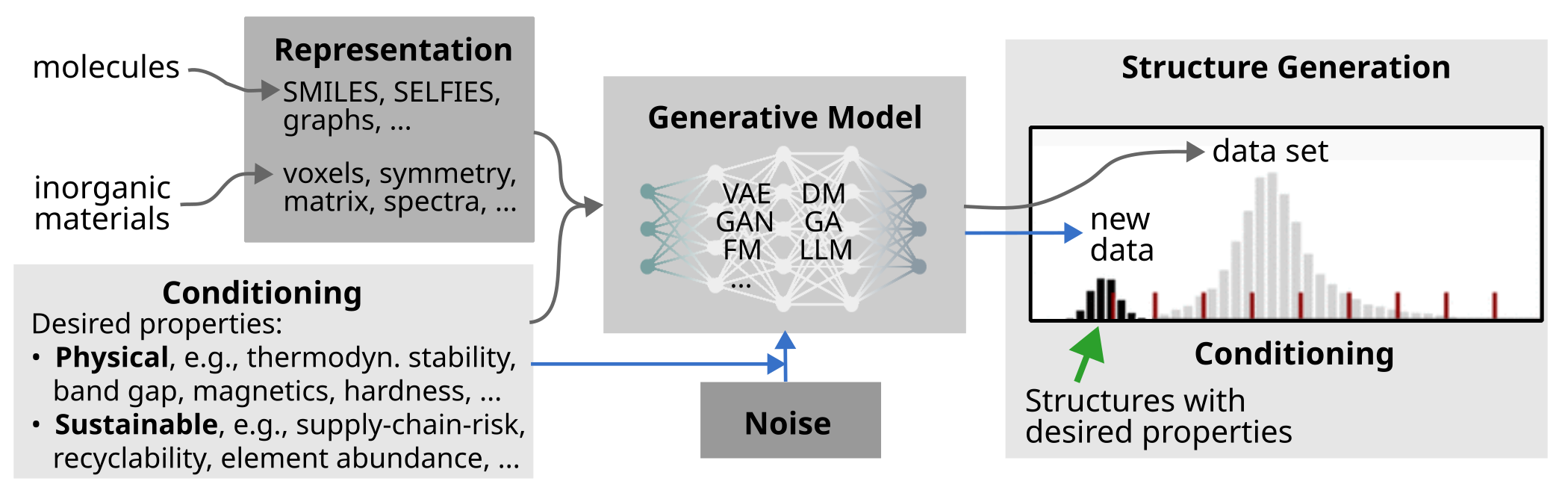
Figure 4: Generative model training and conditional sampling for molecules/materials with desired properties.
AI-Driven PSPP Reasoning and Knowledge Extraction
LLMs are increasingly evaluated for their capacity to encode and reason across the processing-structure-property-performance (PSPP) chain (Figure 5), yet comprehensive, domain-specific benchmarking shows that open, compact, and tailored small LLMs often outperform general-purpose LLMs in both extractive and generative scientific tasks within materials science, particularly when sustainability and interpretability are paramount.
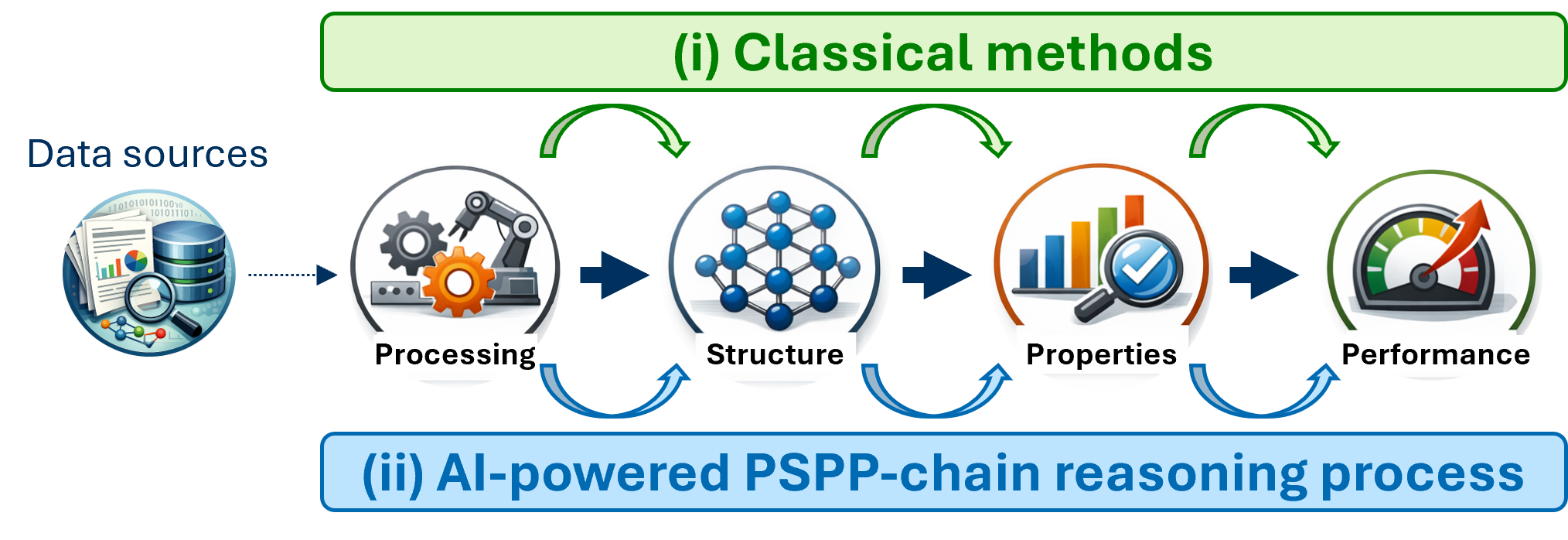
Figure 5: Classical versus AI-powered PSPP workflows in material science.
Extractive BERT-based models trained via knowledge distillation on curated question-answering datasets demonstrate high accuracy, rapid adaptation, and substantially lower carbon costs compared to large generic LLMs. These models excel when integrated as specialized agents in multi-step agentic AI workflows for literature-based data extraction, design-to-device pipelines, and data validation (Figure 6).
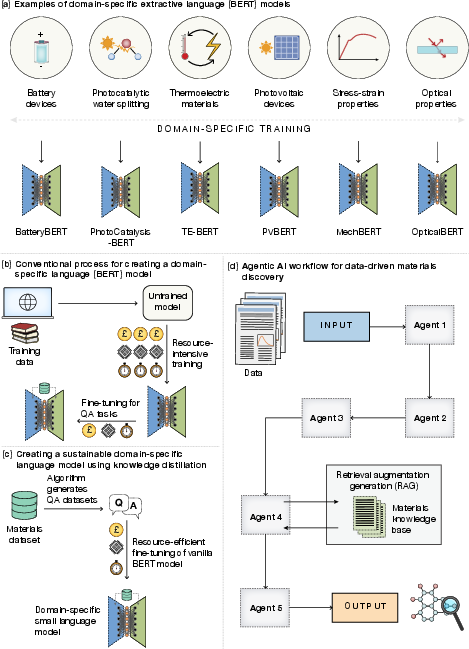
Figure 6: Sustainable domain-specific BERT modeling and integration into agentic AI workflows.
Automated and Active Learning-Driven Sampling
Active learning (AL) methods provide principled, uncertainty-aware sample selection for both MLIP dataset construction and expensive ab initio simulation pipelines, dramatically reducing the computational burden required for model robustification. AL strategies blend structural novelty detection, uncertainty quantification via committees, ensembles, or GP variance, and adaptivity to bring model data efficiency close to optimal.
In large-scale, high-complexity studies (e.g., amorphous, heterostructured, defect-rich, or multi-component systems), BO and multi-task/gradient-augmented surrogate models enable rapid global and local optimization with minimal sample budgets. Iterative and automated workflows (autoplex, pyiron, atomate2, aiida) support reproducible, sustainable data management, and training cycles for both ML and ab initio-based property calculators (Figure 7).
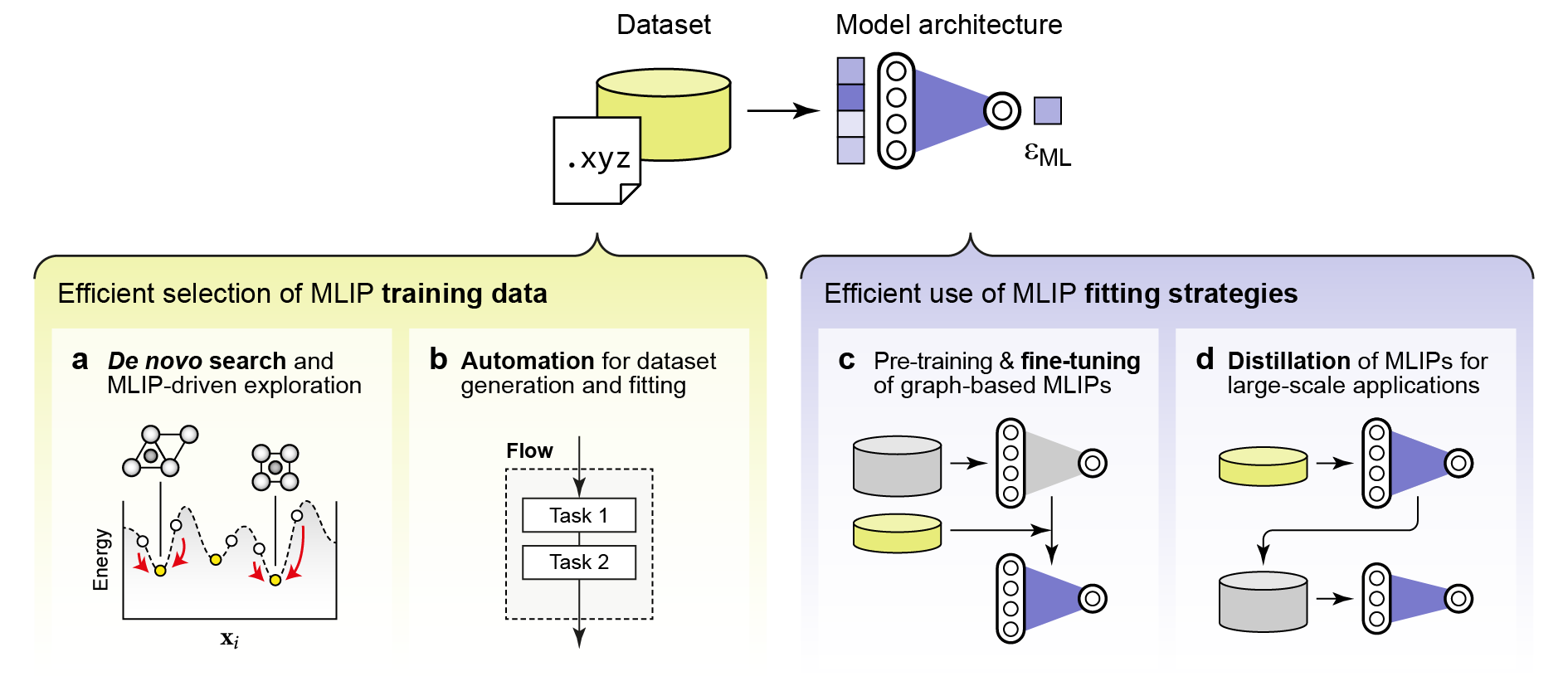
Figure 7: Data efficiency strategies in MLIP training: automated exploration, pre-training, fine-tuning, and model distillation.
High-throughput discovery pipelines have matured to leverage open data, universal models, and modular workflow toolkits. However, remaining bottlenecks for sustainable exploration include the integration of explicit disorder modeling, synthesizability constraints, microstructural effects, and experimental feedback, all of which are essential for practical and scalable advances.
Toward Automated and Autonomous Research
The vision of self-driving laboratories (SDLs) is addressed as an apex of sustainable, efficient exploration where automated workflows, data repositories, and universal surrogate models are orchestrated in an agentic loop from hypothesis generation to experimental validation. Bayesian optimization, coupled with prior knowledge integration, function network-based decision-making, and hierarchical proxy measurement schemes, forms the backbone of data-efficient and objective-driven experimental campaigns.
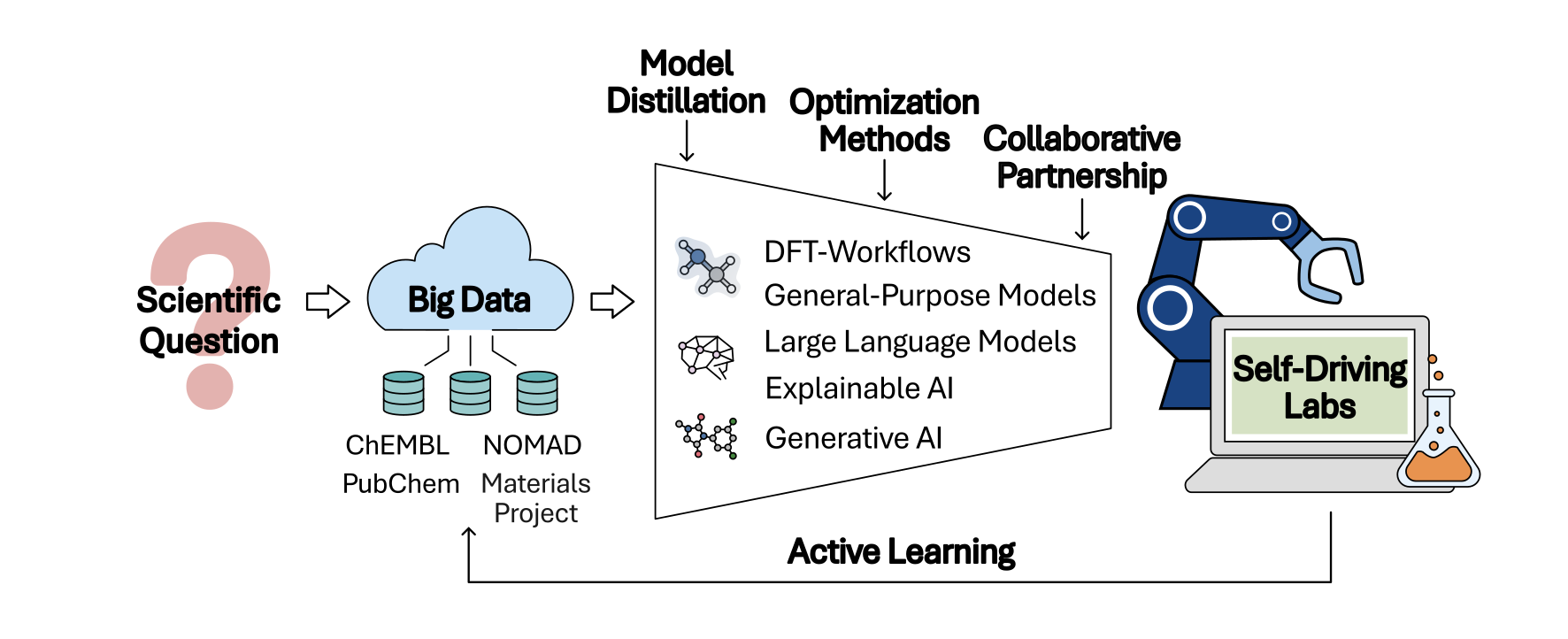
Figure 8: Workflow for sustainable chemical space exploration—self-driving labs enabled by open data, reusable workflows, and scientific partnerships.
The realization of closed-loop, autonomous discovery requires robust integration of synthesis feasibility, uncertainty metrics, and physical constraints at every level of the ML and automation stack.
Conclusions
This perspective establishes a technical roadmap for sustainable machine learning in molecular and materials science by:
- Emphasizing foundational, open, and reusable models and data as critical to amortize the high resource cost of large-scale ML and QM dataset generation.
- Highlighting the necessity of physically grounded, interpretable, and hierarchical ML approaches for data efficiency, generalization, and reliability in complex or sparse-data regimes.
- Demonstrating the centrality of active/automated learning, uncertainty estimation, and modular workflow management for sustainable sampling and scalable automation.
- Recognizing the challenge of realistic model evaluation—incorporating disorder, synthesis accessibility, and real-world property constraints as the next frontier for generative and predictive models.
- Advocating for the development and use of compact, domain-specific AI agents, extractive tools, and knowledge-based workflows to maximize scientific return per unit resource.
Future directions include deeper integration of experimental feedback, physical priors, and agentic AI in fully autonomous research frameworks, especially as disorder, microstructure, and multi-objective optimization become mainstream in computational exploration.
The computational materials and chemistry communities are called to unite around open infrastructure, rigorous benchmarking, and sustainable best practices to ensure that predictive improvements also translate into decreased computational, experimental, and environmental costs in the pursuit of technological and therapeutic materials discovery (2604.00069).

