- The paper establishes a computational framework using Potts models and Monte Carlo optimization to quantify the feasibility of overlapping protein-coding genes.
- It reveals that frame-specific constraints and genetic code degeneracy sharply limit accessible amino acid pairs, with the -2 frame showing up to 49% compatibility.
- The study demonstrates that high-fitness overlapping sequences form connected mutational paths, highlighting practical implications for synthetic biology and genome engineering.
The Fitness Landscape of Overlapping Genes
Introduction
The study addresses a core question in molecular evolution: to what extent can two protein-coding genes be designed or found to overlap within the same DNA sequence in staggered reading frames, given the constraints imposed by both the genetic code and protein fitness landscapes. Overlapping genes are prevalent across the phylogenetic spectrum—including viruses, bacteria, and eukaryotes—yet a comprehensive theoretical framework quantifying when arbitrary protein pairs can be overlapped, and under what constraints, has been lacking. The authors apply Potts-model-based fitness landscapes, robust Monte Carlo optimization, and explicit genetic code shuffling to systematize the exploration, characterize the trade-offs, and define the accessible sequence space for overlapping genes.
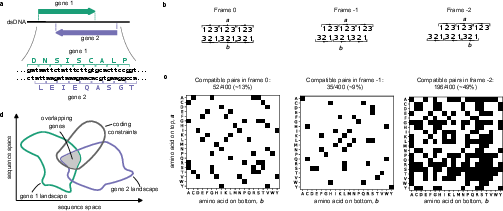
Figure 1: Schematic framework showing reading frames, coding constraints, and the intersection of fitness landscapes for two overlapping genes.
Coding Constraints and Amino Acid Compatibility
The genetic code’s degeneracy (64 codons for 20 amino acids) superficially suggests substantial latitude for encoding multiple proteins in overlapping reading frames. However, detailed enumeration demonstrates severe nonuniformity: in frame 0, only about 13% of all possible amino acid pairs can be mutually encoded; in frame -1, this drops to ~9%; while in frame -2, nearly half (~49%) of all pairs are compatible. These constraints compound exponentially for amino acid doublets. Thus, the actual amino acid sequence space accessible to overlapping genes is highly restricted and fundamentally dependent on frame alignment.
This intrinsic limitation presents a computational satisfiability problem: does the intersection between sequence spaces that support properly folded, functional proteins suffice to allow appreciable overlap for arbitrary protein pairs, or does the fitness landscape become ‘frustrated’ such that overlaps are inaccessible for certain pairs and/or frames?
Potts Model-Based Sampling of Overlapped Sequence Space
Potts models parameterized from multiple sequence alignments offer an empirical proxy for protein family fitness landscapes. The sampling strategy combines the energy functions from two independent Potts models, defining a joint fitness function over the DNA sequence underlying overlapping reading frames. Mutations are introduced at the nucleotide level and are accepted or rejected based on the resulting joint fitness change, with premature stop codons always rejected.
Simulated overlaps of well-characterized folds, including a fibronectin type III domain and a response regulator receiver domain, recover solutions with native-like Potts energies (relative to their MSA-derived means and standard deviations). AlphaFold predictions for maximally overlapping products confirm preservation of characteristic structures, supporting the conclusion that the computational pipeline discovers sequences capable of folding and with likely bona fide activity.
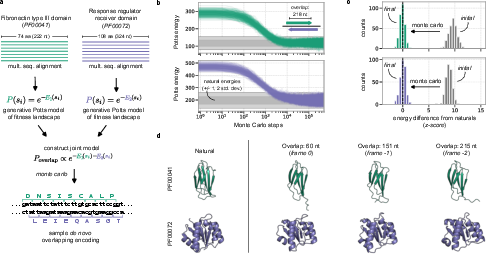
Figure 2: Monte Carlo sampling of overlapping genes using MSA-derived Potts models: convergence of sequence energy, z-score histograms, and structural validation with AlphaFold.
Frustration and Trade-offs in the Joint Fitness Landscape
Not all protein overlaps are feasible, even for moderate overlap lengths. Systematic sampling using replica exchange Monte Carlo (REMC) reveals that for some protein-family pairs and overlap specifications, lowering the fitness penalty for one protein necessarily increases it for the other, indicating frustrated trade-offs. By mapping the Pareto frontier in joint energy space (z-scored), the accessibility of the natural (wild-type) energy regime is directly observable: an overlap is only feasible if the (0,0) z-score—the region occupied by naturally evolved proteins—lies within the joint fitness space. In many cases, and especially at large overlaps or in certain reading frames, this is not achieved.
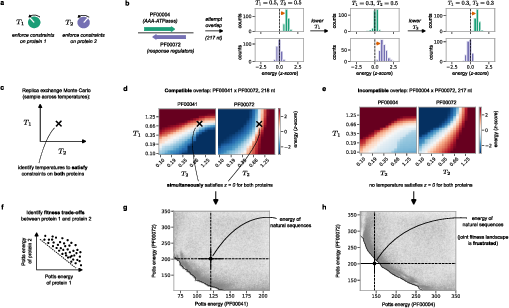
Figure 3: REMC-based analysis identifying compatible and frustrated overlaps, and visualization of accessible energy space and Pareto fronts.
Systematic Survey Across Protein Families and Reading Frames
A broader survey of 17 Pfam protein families (136 unique pairs, multiple overlap lengths, and reading frames) reveals strong, quantitative differences in overlap permissibility depending on reading frame. Frame -2 is quantitatively more permissive, supporting maximal overlaps (up to ~300 nt) in a majority of protein pairs, with >90% success rates for moderate overlaps. In contrast, frames 0 and -1 rapidly lose compatibility past 150–200 nt of overlap, especially for functionally and structurally divergent pairs. The high permissiveness of the -2 frame is explained by codon structure: the third (‘wobble’) position of one frame aligns with the second (most constrained) position of the other, maximizing encoding flexibility.
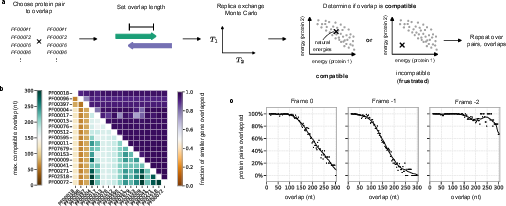
Figure 4: Compatibility matrix for maximal feasible overlaps across protein family pairs, as a function of reading frame and overlap length.
The Role of the Genetic Code’s Architecture
To interrogate whether the standard genetic code is uniquely supportive of overlapping genes, two shuffling schemes are examined: (i) per-codon reassignment preserving degeneracy (type I); (ii) synonymous block relabelling (type II). Both randomization schemes reduce, but do not abolish, overlap feasibility. However, in all cases, the standard code enables more accessible, lower-energy overlap solutions, particularly in frame -2. The architecture of the standard code thus confers an explicit flexibility with respect to overlapping gene evolution not fully explained by degeneracy or synonymous mutation structure alone.
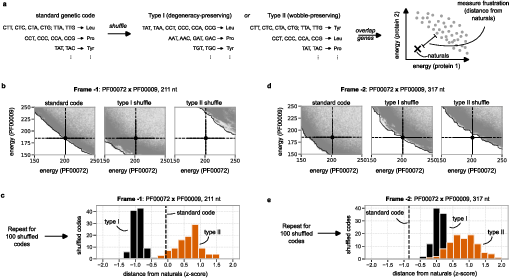
Figure 5: Comparison of Pareto front accessibility in the standard genetic code versus random degenerate and synonymous-block shuffled codes.
Connectivity in Sequence Space
Evolutionary accessibility requires not only the existence of feasible overlaps but also that the set of high-fitness overlapping sequences is mutationally connected. Pairwise sequence ‘endpoints’ (with substantial sequence divergence) are connected by optimized mutational paths, identified via genetic algorithms, with all intermediates maintaining ∣z∣<0.25 for both proteins. This demonstrates that the space of overlapping solutions, while strictly reduced compared to non-overlapping genes, remains navigable by single-nucleotide steps within the fitness plateau as simulated.
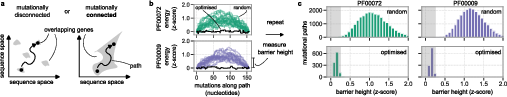
Figure 6: Mutation path analysis showing that independently-generated overlapping genes can be connected by functional intermediates.
Implications and Future Directions
This work provides computational evidence that the intrinsic architecture of the genetic code, notably its degeneracy and synonymous patterning, enables a substantial but sharply delimited region of sequence space for overlapping protein-coding genes. The -2 reading frame emerges as unexpectedly privileged for such overlaps, with practical implications for synthetic biology, viral genome engineering, and de novo gene design. The mutational connectivity result suggests that, in principle, protein-coding overlaps do not represent evolutionary dead-ends—sequences are embedded in a connected, albeit sparser, fitness landscape.
The study reveals that certain genetic code architectures (e.g., natural versus randomized) are non-trivially better suited to overlapping; this may inform theories of code evolution and the constraints acting on biosynthetic code expansion.
The approach is limited by reliance on Potts-model fitness approximations, which capture local but not higher-order epistatic effects, and experimental validation will be required to systematically benchmark synthetic deep overlaps. Future developments should integrate higher-order LLMs, empirical fitness data, and explore overlapping protein-coding design in real cellular contexts.
Conclusion
The formalism unifies the quantification of coding and functional constraints on overlapping genes, demonstrates a broad range of compatibility for protein pairs—driven by reading frame and genetic code properties—and underscores the evolvability of such architectures via connected mutational networks. These results advance theoretical understanding of protein-coding overlap, with implications for bioinformatics, genome annotation, and synthetic genetic system engineering.