- The paper introduces the RealMat-BaG benchmark that rigorously evaluates ML models on experimental bandgap data using integrated computational and curated experimental datasets.
- The study shows that computational pretraining enhances convergence and robustness on limited experimental data, though it may worsen predictions in challenging out-of-distribution chemical domains.
- Interpretability analyses reveal that chemically intuitive motifs drive predictions and that classical models can perform competitively with advanced graph neural networks.
Benchmarking Machine Learning for Realistic Experimental Bandgap Prediction
Introduction
Accurate prediction of electronic bandgaps is central to the computational design and assessment of semiconductors for optoelectronic and energy materials. Despite substantial algorithmic progress, a gap persists between ML models trained on computational datasets and their predictive reliability under experimentally relevant conditions. The paper "Benchmarking bandgap prediction in semiconductors under experimental and realistic evaluation settings" (2604.25568) introduces the RealMat-BaG benchmark, specifically designed to rigorously evaluate ML models on experimental bandgap data, probe their robustness to domain and distributional shifts, and systematically investigate questions of transfer and interpretability that are currently underexplored in the field.
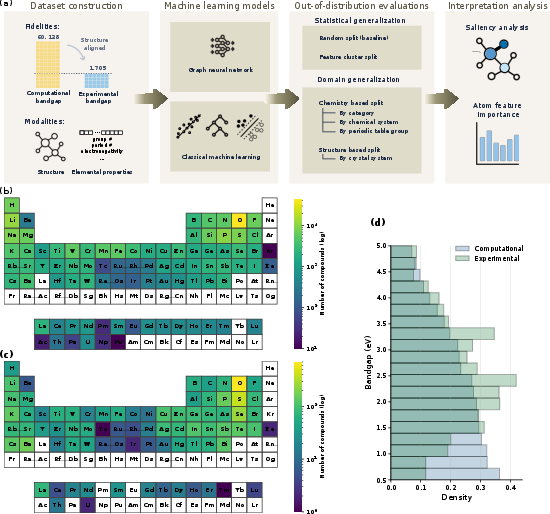
Figure 1: Overview of the RealMat-BaG benchmark design, integrating computational and experimental bandgap datasets with respective crystal structures, and assessing models over in-distribution, feature-based, and domain-based OOD splits, with interpretability analyses via graph-based attribution and SHAP.
Benchmark and Dataset Composition
RealMat-BaG consists of two integrated components: a large-scale DFT-PBE computational dataset from the Materials Project, and a meticulously curated experimental bandgap dataset, the latter being aligned with explicit crystal structures. The experimental split is unique both in its assembly—drawing from validated literature sources and enforcing traceability through DOI linking—and in its extensiveness, comprising over 1,700 entries across diverse chemistries. Both datasets are systematically analyzed to expose gaps in compositional and target space: while the computational dataset spans the vast majority of the periodic table, the experimental data are notably denser in chalcogenides and sparser for heavy metals and other less-accessible compounds. Importantly, significant differences exist between the computational and experimental ground-truth distributions, with experimental bandgaps clustering in a narrower, higher-fidelity range.
Evaluation Protocols and OOD Generalization
Four complementary evaluation regimes are defined: (i) random splits for traditional in-distribution generalization, (ii) feature-based distributional shifts obtained via unsupervised clustering, (iii) domain-based chemistry-aware splits (e.g., by material category, chemical system, periodic group), and (iv) structure-based OOD splits at the crystal system level. These enable rigorous quantification of model robustness well beyond random cross-validation.
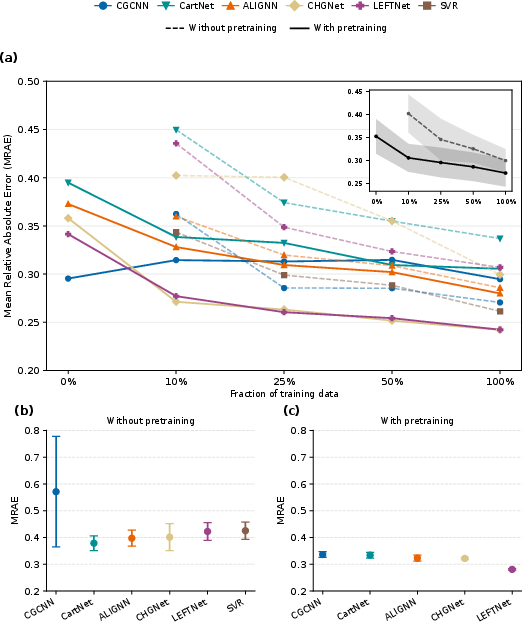
Figure 2: Pretraining on computational data enhances data efficiency and stability, especially when experimental supervision is limited; models initialized with computational weights outperform those trained from scratch for small data fractions and under feature-based OOD settings.
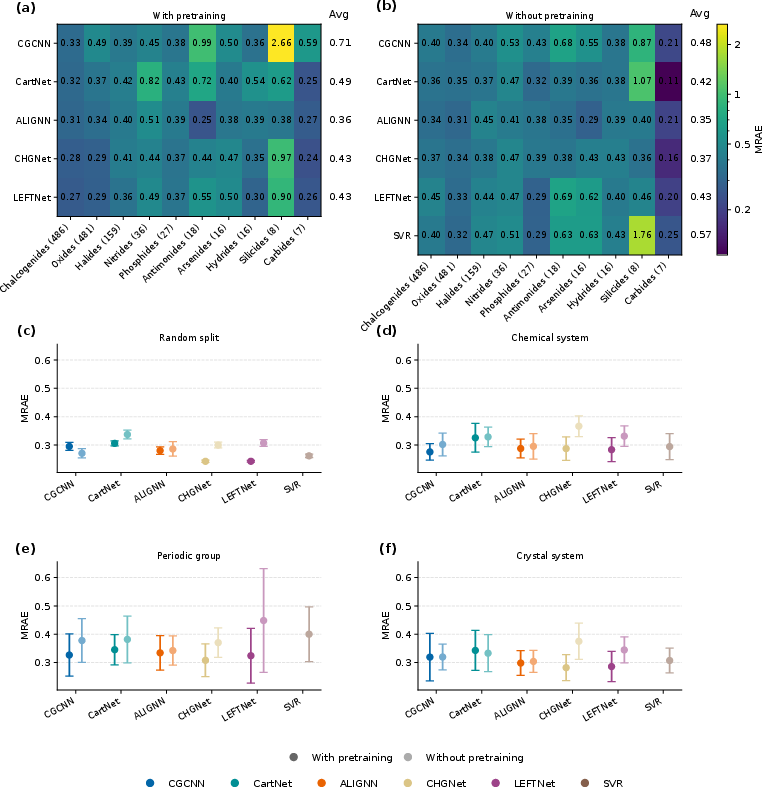
Figure 3: Large performance degradation is observed for all models under domain-based OOD protocols, with outliers such as silicides and antimonides consistently showing the highest MRAE, reflecting a fundamental limit in cross-domain extrapolation.
Results indicate that random splits systematically overestimate generalization—MRAEs in random settings are consistently lower (typically 0.24–0.30 for SOTA GNNs), while domain-based OOD splits yield significantly higher errors (MRAE frequently >0.5 for challenging categories). The silicides and antimonides families represent particularly recalcitrant domains, where all model classes—regardless of pretraining or input encoding—struggle and where pretraining can even increase error, highlighting the propagation of computational dataset biases.
Influence of Pretraining and Model Family
The effect of computational pretraining is nuanced. For most modern GNNs (ALIGNN, CHGNet, LEFTNet), pretraining accelerates convergence and improves robustness under limited experimental data; the effect, however, diminishes as experimental supervision increases. Not all architectures benefit—CGCNN, for instance, occasionally achieves lower MRAE when trained exclusively with experimental data, without any pretraining. This effect is visible both in random and OOD settings, and is material- and architecture-dependent. Most notably, pretraining can be detrimental in certain domains, amplifying systematic DFT errors.
Atomic Encoding, Inductive Bias, and Interpretability
The benchmark explores the effect of atomic encoding—atomic number (Z) vs. property-based (Prop)—on both accuracy and interpretability. While Prop encodings can improve LOMO category-level OOD generalization (particularly for silicides/antimonides), the benefit is nonuniform and context-dependent, revealing that chemically informed priors alone are insufficient to fully close domain gaps.
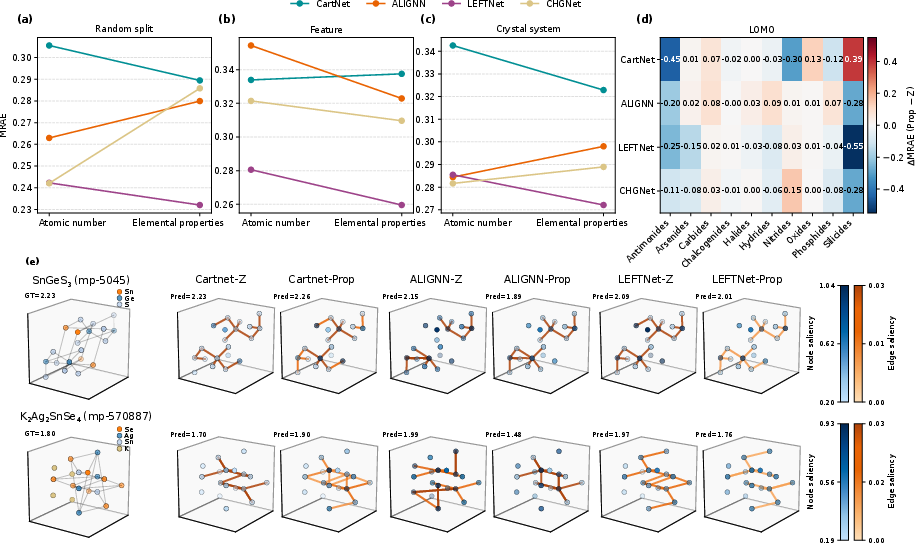
Figure 4: Prop encodings deliver marginal or task-specific improvements over Z encodings; gradient saliency maps across representative models consistently highlight physically meaningful atomic and bonding motifs as most critical for bandgap prediction.
Saliency analyses across CartNet, ALIGNN, and LEFTNet show convergent emphasis on chemically intuitive motifs—chalcogen-metal bonds, valence-linking atoms—corroborating the physical plausibility of learned representations, especially in models encoding higher-order geometry. In parallel, classical models (notably SVR) are revealed to be strong baselines, in some circumstances surpassing GNNs and offering outstanding interpretability, though at the cost of explicit geometric expressivity.
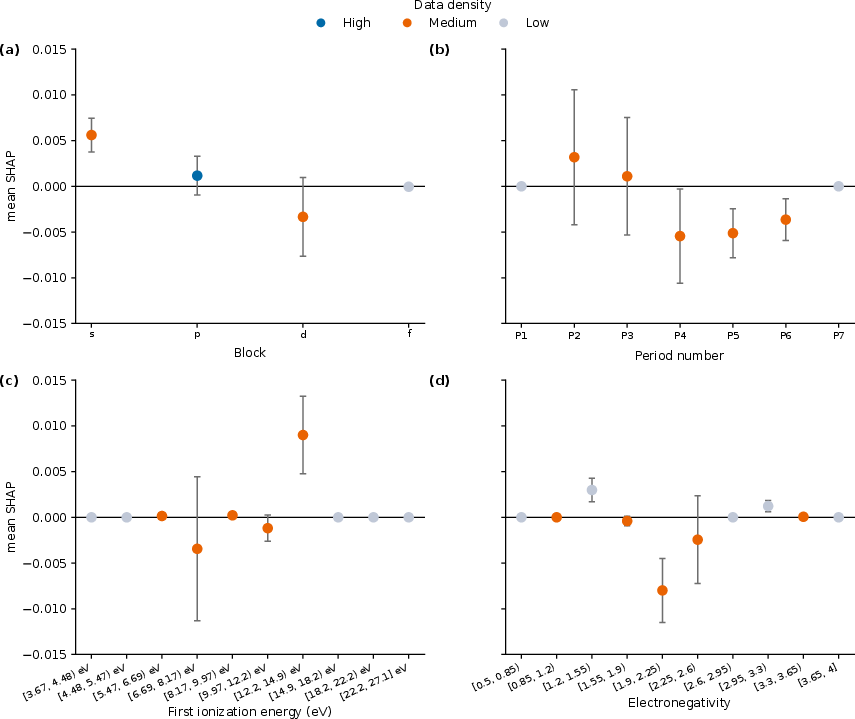
Figure 5: SHAP analysis for SVR reveals that d-block content and higher period numbers systematically correlate with smaller bandgaps, consistent with established solid-state chemistry, while feature importance also reflects dataset composition effects.
Limitations and Impact
Despite progress, all models—DL and classical—exhibit pronounced failures for difficult chemical domains and under abrupt distributional shifts. The reported MRAEs, exceeding 0.5 in key OOD cases, are substantial enough to affect material screening and downstream decision-making. These errors are shown to be structural, not merely algorithm-dependent. The analysis makes clear that transfer and generalization are bottlenecked not by representation richness alone but by a combination of data fidelity, compositional coverage, and the extent to which experimental complexity is physically encoded.
Practical and Theoretical Implications
The RealMat-BaG benchmark evidences that (1) computational pretraining can be highly beneficial for data-limited scenarios but is not a panacea for OOD generalization, (2) domain-aware splits are necessary for realistic appraisal of model deployment reliability, and (3) careful interpretability analysis is essential for establishing trust in model-guided synthesis. The strong performance of classical models suggests that much predictive signal resides at the aggregate property level, but explicit modeling of local geometric environments remains necessary.
From a theoretical standpoint, the findings underscore the importance of benchmarking on experimental data with standardized protocols and of developing future multi-fidelity and multimodal learning paradigms that directly encode extrinsic information such as synthesis conditions, defects, and phase distributions.
Conclusion
RealMat-BaG defines the current state-of-the-art in benchmarking ML for experimental materials discovery, setting a high bar for model reliability, domain robustness, and interpretability. The framework exposes key limitations in out-of-distribution generalization—limitations that cannot be resolved via pretraining or architectural changes alone—and establishes a path toward more trustworthy deployment of ML in physical sciences. Future directions involve enriching datasets with greater metadata, adopting more nuanced OOD definitions, and ultimately developing models that capture the full complexity of experimental conditions for bandgap prediction and beyond.