- The paper introduces an RL-guided combinatorial chemistry framework that overcomes traditional models' limitations in extrapolating material properties.
- It integrates BRICS-based molecular fragmentation with proximal policy optimization and action masking to efficiently generate target molecules.
- Empirical validation shows the method's success in discovering superior protein docking compounds and potent HIV inhibitors.
Overview of Reinforcement Learning-Guided Combinatorial Chemistry for Materials Discovery
The research paper titled "Materials Discovery with Extreme Properties via Reinforcement Learning-Guided Combinatorial Chemistry" (2303.11833) introduces an approach to leverage reinforcement learning (RL) for combinatorial chemistry, aiming to discover new materials with properties that exceed those currently known. This method addresses the limitations of traditional machine learning models, particularly those that rely on probability distribution learning, which have proven insufficient for discovering materials outside the scope of their training data distribution.
Theoretical Limitations of Probability Distribution-Learning Models
Inverse molecular design models such as NMT, VAE, and GAN typically learn the empirical probability distribution of the training data, denoted as Pdata. These models are structured to approximate this distribution rather than the true probability distribution of the chemical systems being studied.
- NMT Models: These models maximize the likelihood of matching input and output sequences of molecular structure and properties. The learning process involves approximating the hypothesis to the empirical probability distribution through the minimization of cross-entropy, which inherently limits their capability for materials extrapolation.
- VAE Models: These models constrain latent variables to follow a prior distribution like a normal distribution and use a reconstruction error term akin to negative log-likelihood, ultimately learning an approximation to Pdata.
- GAN Models: GANs are trained to generate data resembling the training set to deceive a discriminator, and at optimal conditions, they achieve this by learning Pdata as opposed to true material distributions.
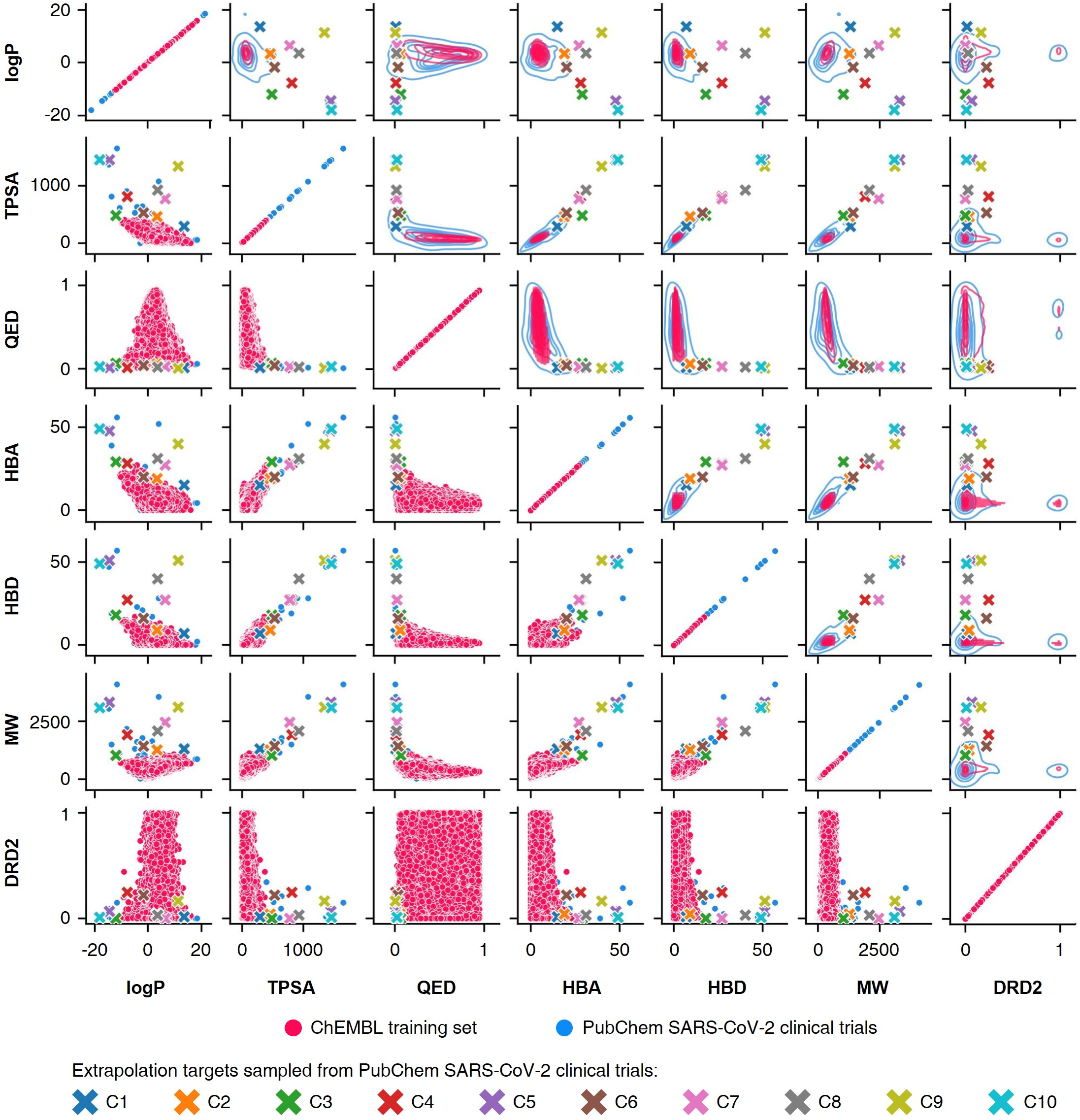
These models' reliance on empirical data distributions makes them unsuitable for materials extrapolation challenges where novel compounds with unprecedented properties must be discovered (Figure 1).
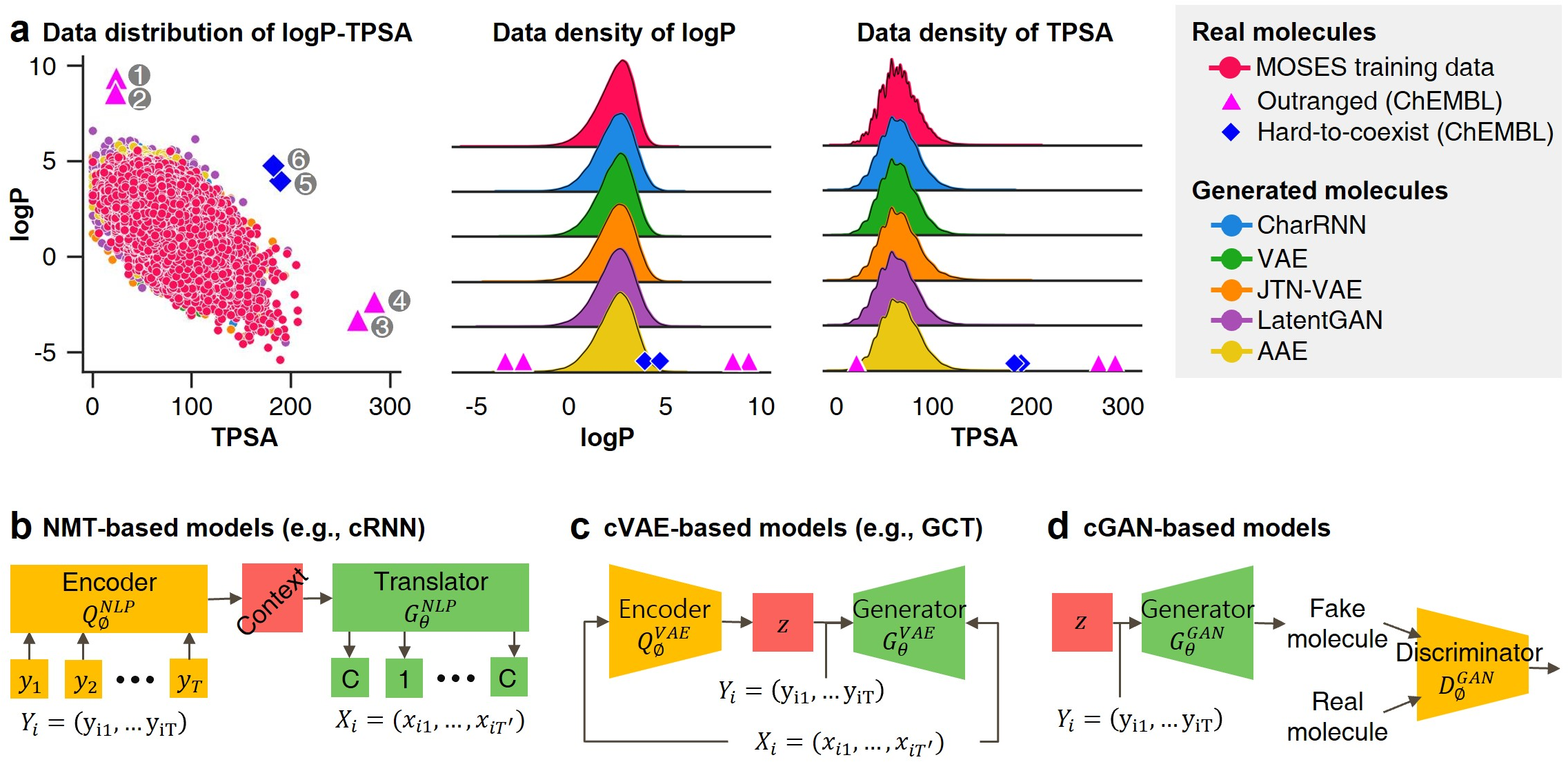
Figure 1: Probability distribution-learning models for molecular generation. Data distribution illustrates the challenges in extrapolating beyond known data.
RL-Guided Combinatorial Chemistry Framework
The RL-guided combinatorial chemistry method integrates a learning policy to select promising molecular fragments that combine into desired molecules. This approach essentially consists of:
- Configuration Settings: Involves defining task types (specific values vs. maximization of targets), reward functions, termination, and target conditions. The fragmentation process is guided using BRICS rules (Figure 2).
- Training Phase: Utilizes the proximal policy optimization (PPO) for policy learning. Action masking is applied to improve learning efficiency by reducing action space to feasible fragment combinations.
- Inference Phase: Demonstrates the generation of molecules using the learned policy, ensuring that the resultant molecules meet the designed targets (Figure 3).
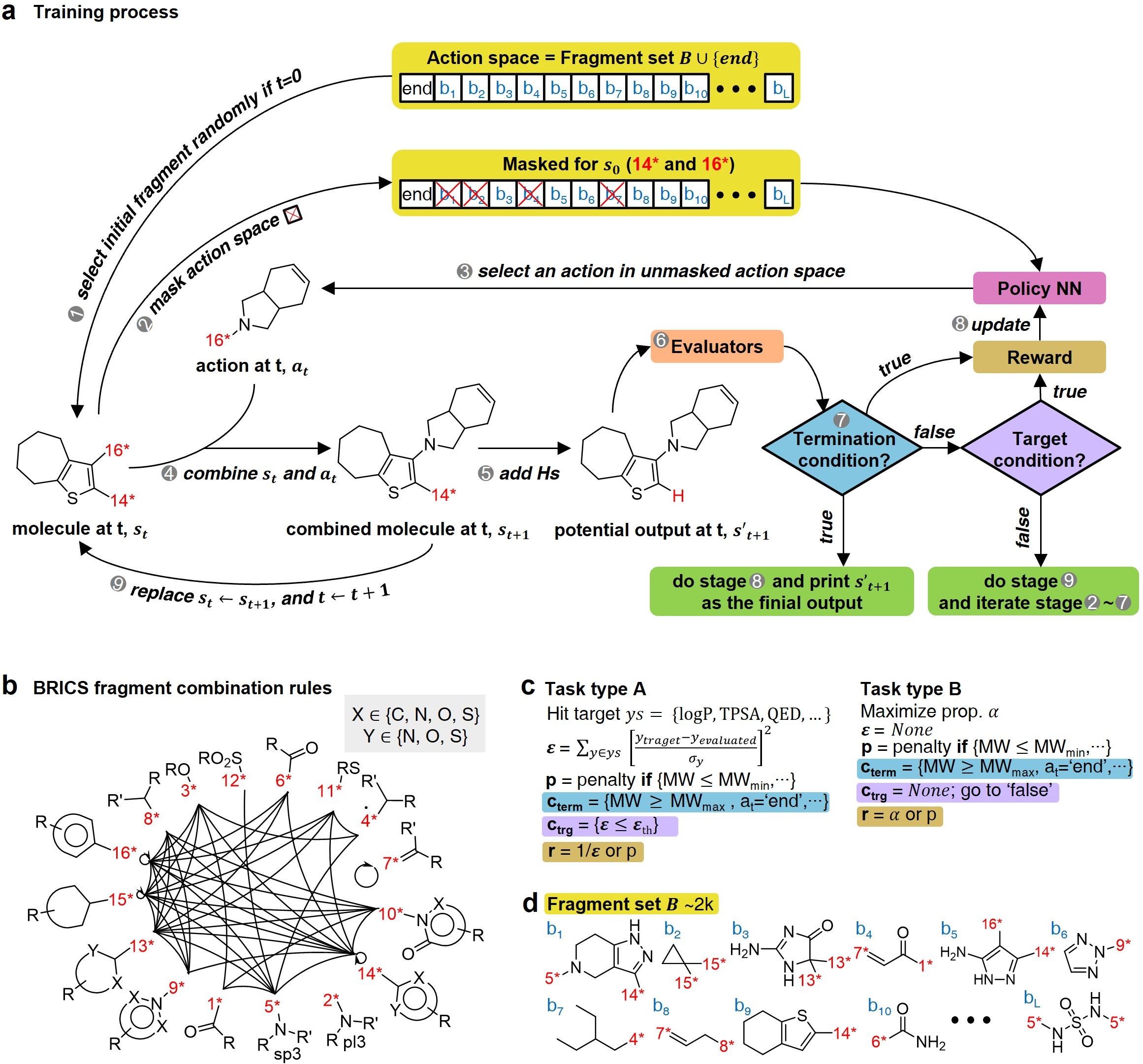
Figure 2: Overview of RL-guided combinatorial chemistry with BRICS, showcasing model training and task-setting methodologies.
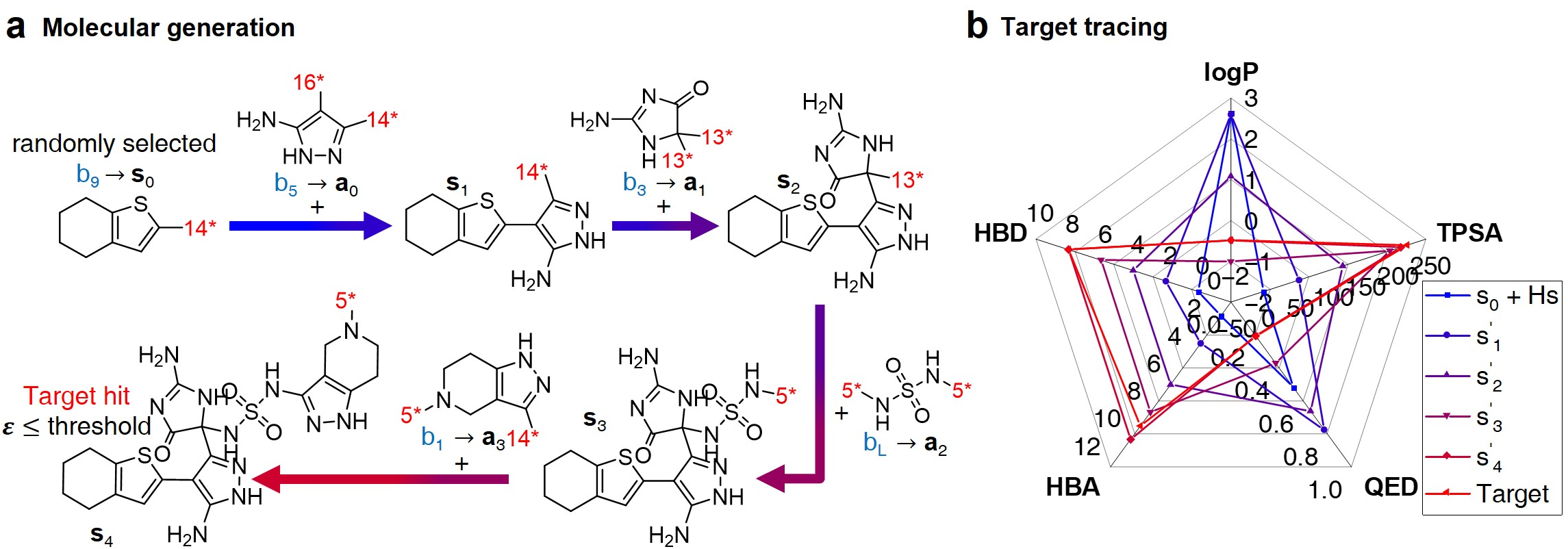
Figure 3: Inference process for molecular generation, demonstrating property changes throughout fragment combination.
The paper empirically verifies the capability of RL-guided combinatorial chemistry in successfully discovering molecules with extreme properties. Notably, in comparison to probability distribution-learning models such as cRNN and GCT:
Practical Applications
The RL-guided approach was practically implemented to discover:
- Protein Docking Molecules: Targeting the 5-HT\textsubscript{1B} receptor, RL-guided combinatorial chemistry outperformed traditional methods in identifying molecules with superior docking scores, confirming its practical utility in drug discovery (Figure 5).
- HIV Inhibitors: The methodology led to significant outperformance in generating inhibitors with high predicted pIC\textsubscript{50} values for targets CCR5, INT, and RT, representing a valid application in pharmaceutical development (Figure 6).
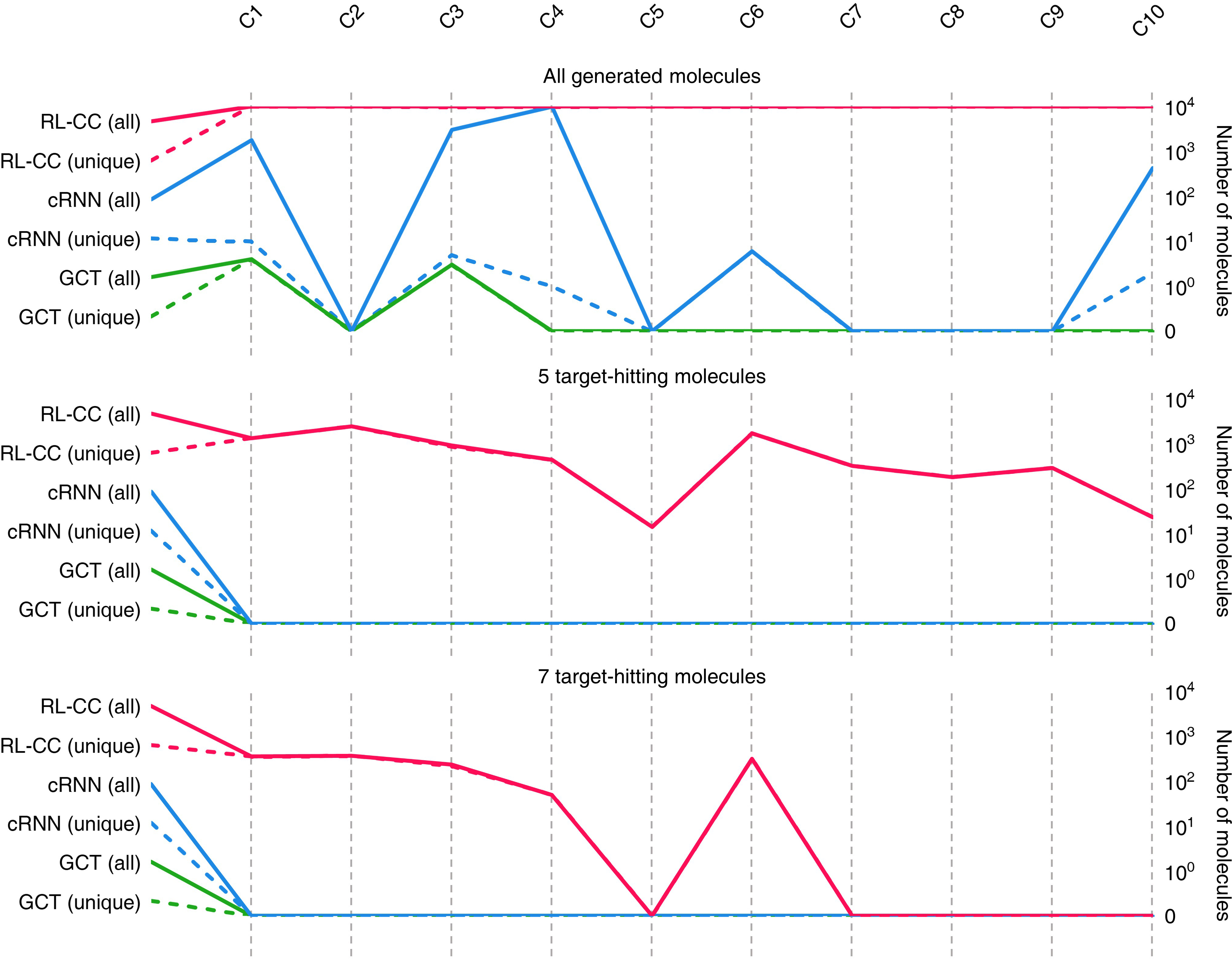
Figure 5: Quality benchmarks of generated molecules showcasing 5-HT\textsubscript{1B receptor docking affinities.
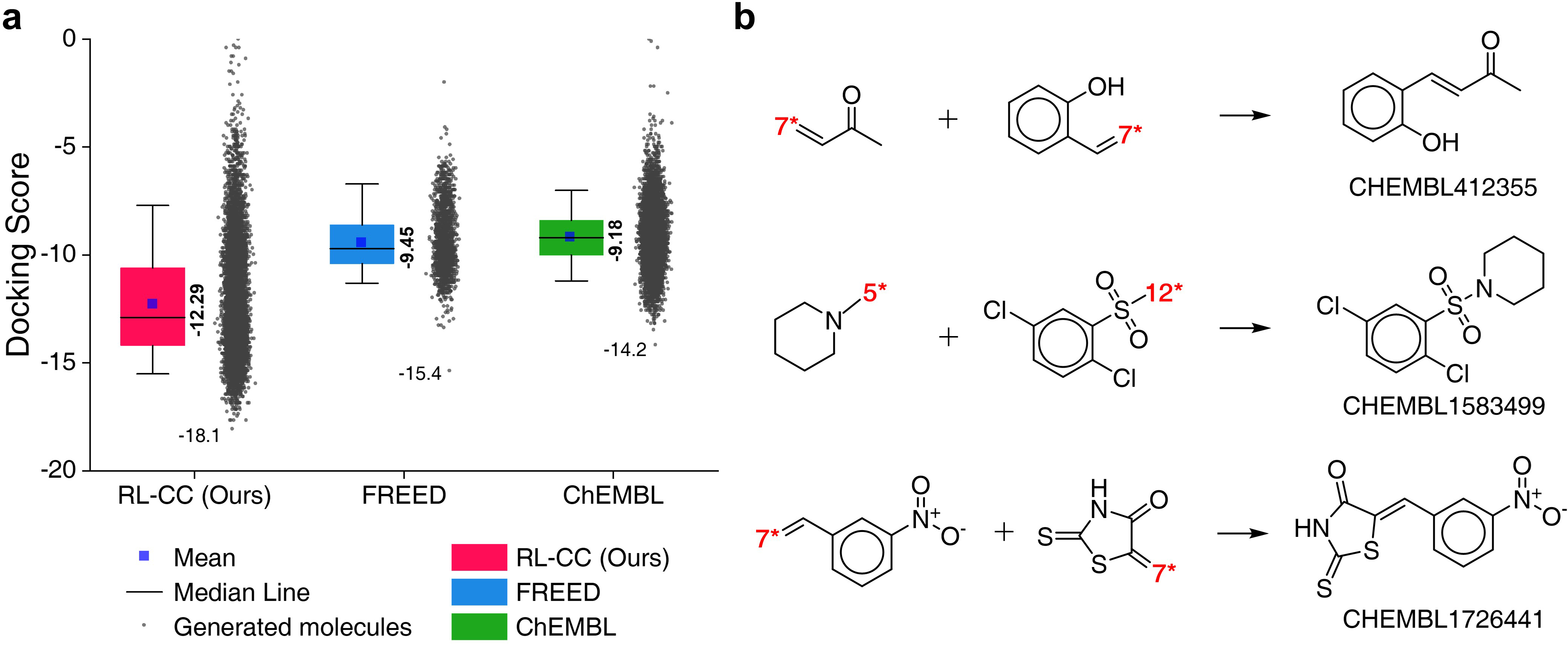
Figure 6: Results for HIV inhibitors discovery, reflecting generation success with high pIC\textsubscript{50 values.
Future Potential and Conclusion
The study concludes that RL-guided combinatorial chemistry has broad applicability beyond drug discovery, potentially extending into organic materials due to its flexible design of molecular fragments using BRICS rules. However, limitations, such as the need for retraining for novel targets and sparse rewards, suggest areas for further study, including the application of meta-learning and hierarchical reinforcement learning techniques for model improvement.
Overall, this paper presents significant advances in the field of materials discovery by leveraging RL to address prior limitations of existing machine learning approaches, confirming the model's ability to extrapolate and discover materials with extreme properties effectively.