- The paper demonstrates that the SQD method efficiently computes CH2 singlet‐triplet dissociation energies with deviations of only a few milli-Hartrees compared to classical methods.
- It integrates classical PySCF computations with quantum LUCJ circuits on a 52-qubit system, employing error mitigation strategies like dynamical decoupling to reduce noise.
- The study highlights SQD’s potential in simulating open-shell systems, which can advance research in combustion and interstellar chemistry.
Quantum-Centric Study of Methylene Singlet and Triplet States
Introduction
The research titled "Quantum-Centric Study of Methylene Singlet and Triplet States" (2411.04827) investigates the CH2 molecule using advanced quantum computing techniques. The study employs a 52-qubit quantum system to calculate the dissociation energies for both the ground state triplet and the first excited state singlet using the Sample-based Quantum Diagonalization (SQD) method. This approach is significant for interstellar and combustion chemistry. Importantly, this work pioneers the application of SQD to an open-shell system like the CH2 triplet, addressing the complex challenges posed by such systems.
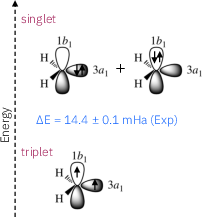
Figure 1: Highest occupied molecular orbitals of methylene, along with the corresponding electronic configurations for the ground state triplet and the first excited state singlet.
Methodology
The methodology encompasses both classical and quantum computational approaches. Classical computations serve as a reference, utilizing the PySCF package with correlation-consistent cc-pVDZ basis sets. The quantum computations employ SQD within a Quantum-Centric Supercomputing framework, effectively simulating electronic structures with large-scale quantum hardware.
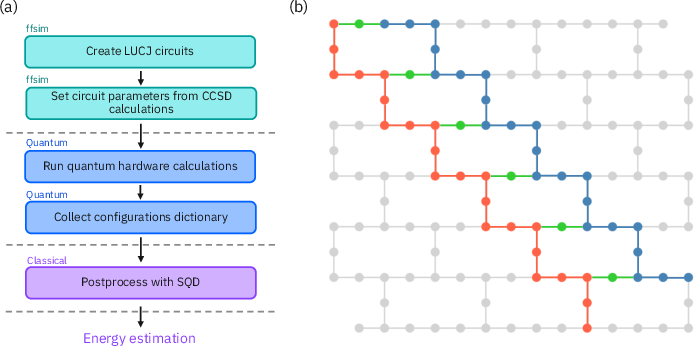
Figure 2: Schematic representation of the concerted approach workflow and qubit layouts for methylene simulations.
The quantum portion involves executing LUCJ circuits on IBM's 52-qubit setup to encode the electronic configurations and explore the energy landscape efficiently. The methodology integrates error mitigation strategies such as dynamical decoupling and gate twirling to minimize quantum noise impacts.
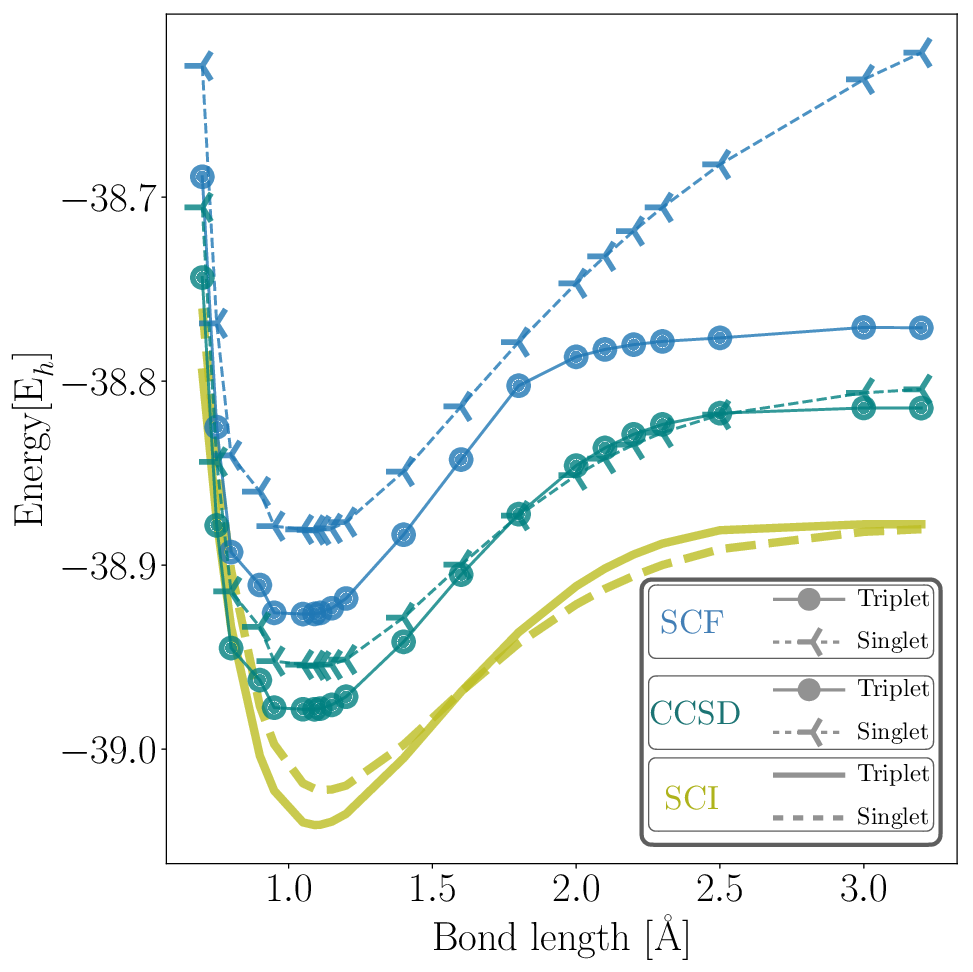
Figure 3: Classical calculations for CH2 singlet and triplet dissociation profiles using cc-pVDZ basis set.
Results and Discussion
The results underscore the capability of the SQD method to rival classical techniques such as Selected Configuration Interaction (SCI) and Coupled Cluster Singles and Doubles (CCSD). Notably, the SQD-derived singlet-triplet energy gap aligns closely with both experimental data and SCI calculations, typically deviating within a few milli-Hartrees (mEh).
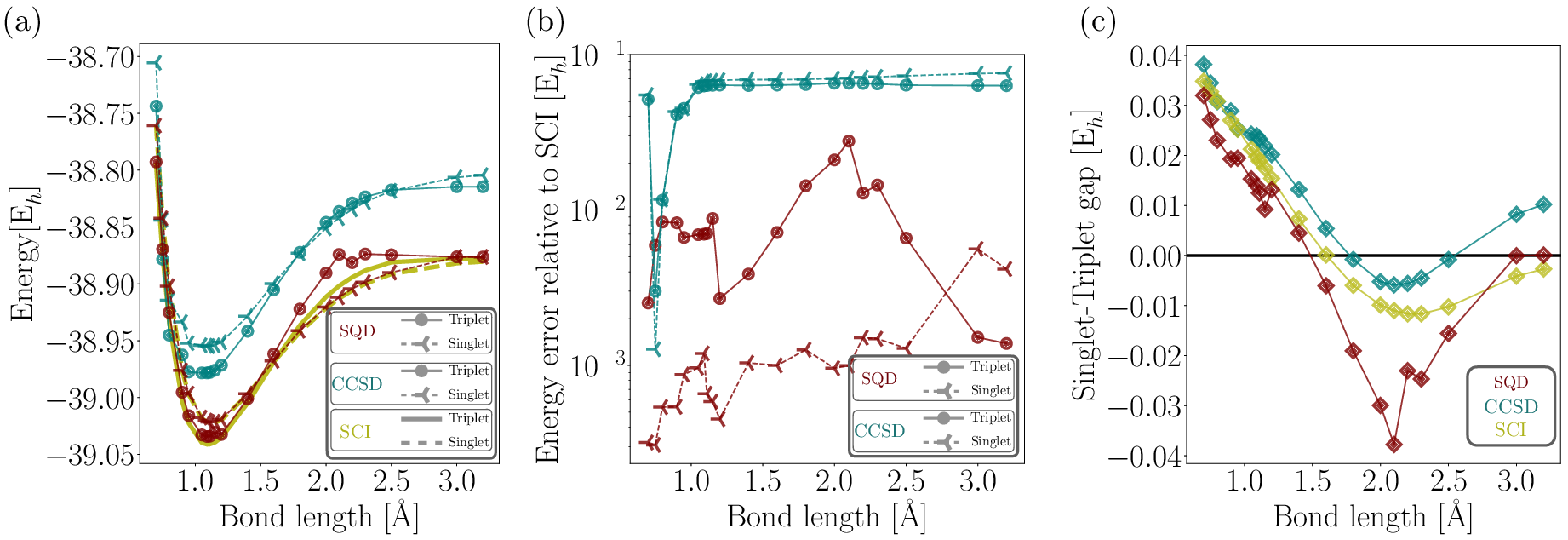
Figure 4: CH2 singlet and triplet dissociation energies along the PES for bond lengths ranging from 0.75 \text{Å} \text{ to } 3.20 \text{Å}, calculated using SQD, CCSD, and SCI.
Variability in the triplet state energies is attributed to complex wavefunction characters and bit-string handling in open-shell systems. Despite these challenges, SQD demonstrates substantial potential in accurately modeling these states, outperforming classical methods, especially in scenarios with significant electron correlation.
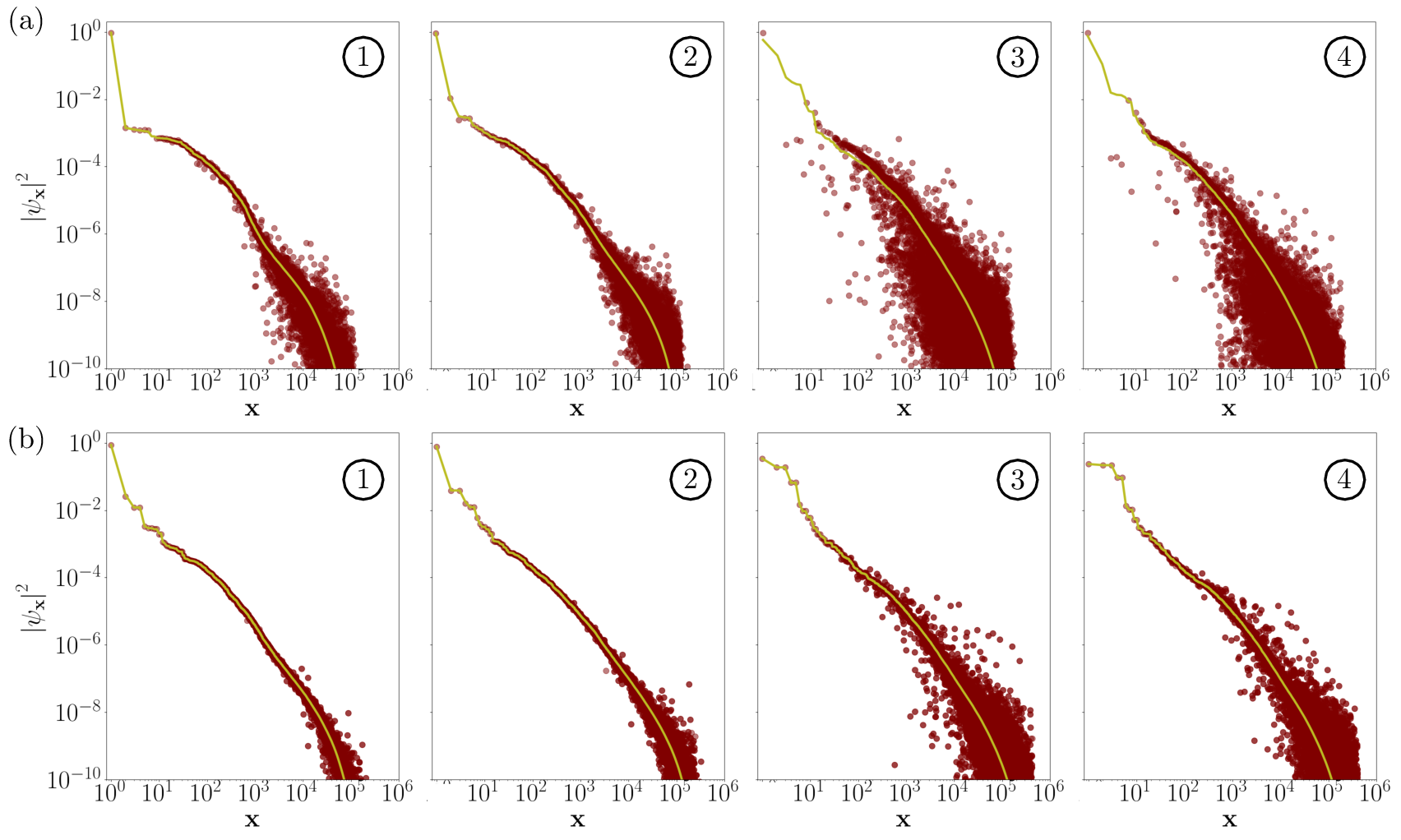
Figure 5: Comparison of the SCI and SQD wavefunction amplitudes for all electronic configurations.
Conclusion
This study advances the understanding and application of quantum algorithms in computing electronic structures of open-shell systems. Using SQD, the study achieves significant agreement with classical and experimental results, marking a milestone in quantum computing for chemical applications. Future research will likely enhance SQD's accuracy in diverse chemical systems, expanding quantum computing's relevance in electronic structure prediction and molecular dissociation studies. The potential for SQD to improve modeling of complex reactions and transient species could significantly impact fields such as combustion chemistry and materials science.
