- The paper presents a novel integration of entanglement forging and sample-based quantum diagonalization to enhance the simulation of hydrogen abstraction reactions.
- The study employs a hybrid quantum-classical framework that reduces qubit requirements while achieving activation energies comparable to gold-standard CCSD methods.
- The approach is validated against DMRG benchmarks, indicating its potential to advance quantum chemical simulations of material degradation in aerospace engineering.
Quantum-centric Simulation of Hydrogen Abstraction
Introduction
The paper "Quantum-centric simulation of hydrogen abstraction by sample-based quantum diagonalization and entanglement forging" presents a novel approach to simulating radical chain reaction pathways using quantum computing. The integration of sample-based quantum diagonalization (SQD) and entanglement forging (EF) enables efficient computation of electronic configurations in radical chain reactions, particularly highlighting their applicability in understanding the photodegradation processes of composite resins used in aerospace engineering. This simulation primarily focuses on hydrogen abstraction reactions, a critical step in the photo-oxidation of epoxy resins.
Methodological Developments
The study leverages a combination of EF and SQD methodologies within a quantum-centric supercomputing (QCSC) framework, wherein a quantum processor cooperates with classical high-performance computing (HPC) resources. The entanglement forging technique significantly reduces qubit requirements by mapping spatial orbitals to qubits, facilitating larger scale simulations with limited quantum resources. EF's core feature is representing a $2M$-qubit electronic wavefunction with multiple M-qubit circuits, which is both a reduction in complexity and resource utilization.
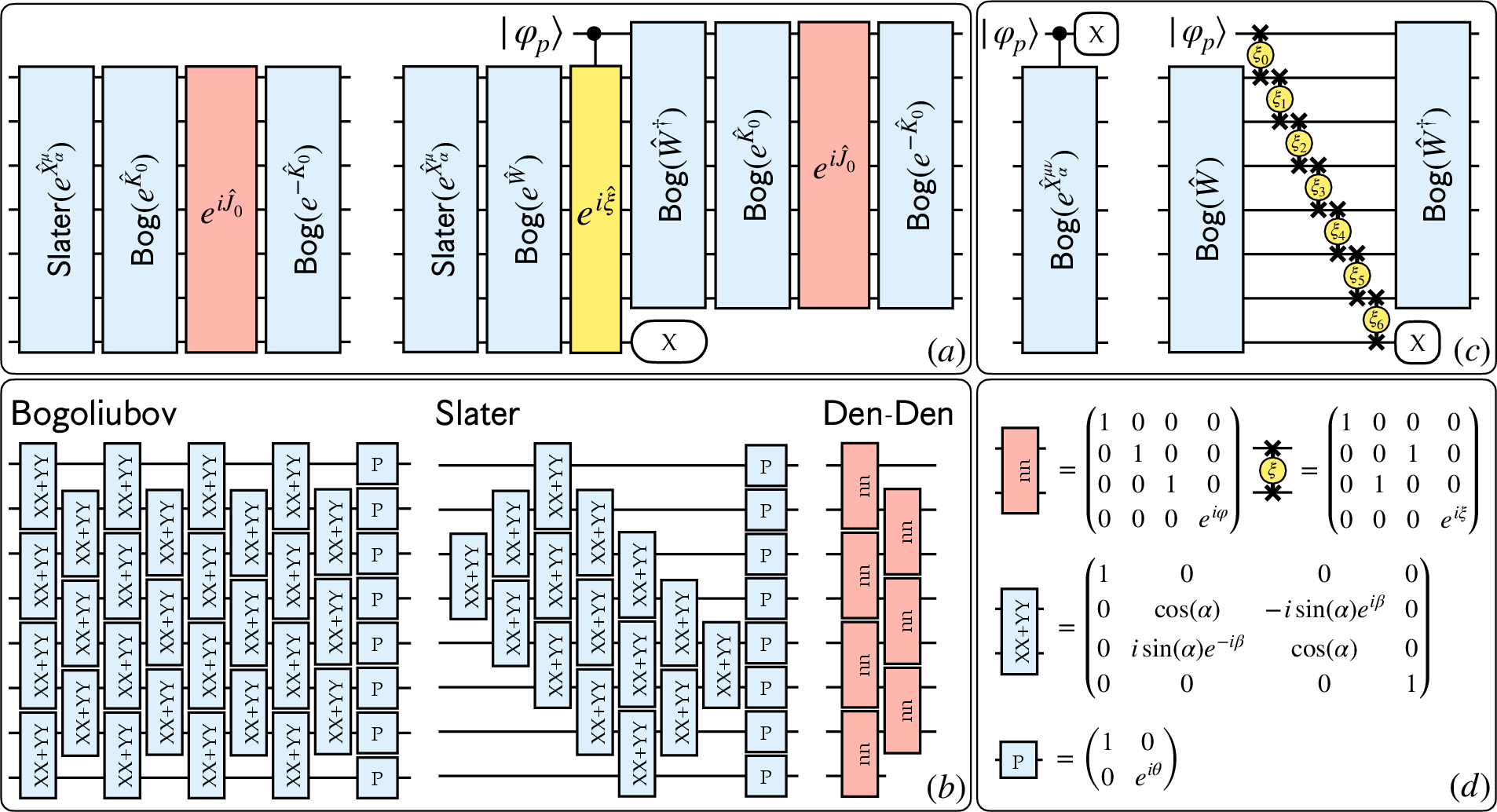
Figure 1: Quantum circuits to prepare the states uμ (left) and uμν (right).
Results of Hydrogen Abstraction Reaction Simulation
The paper details simulations of hydrogen abstraction reactions using 2,2-diphenylpropane as a model, representing epoxy resin's degradation pathway in aerospace materials (Figure 2). The authors observed remarkable accuracy in predicting activation and reaction energies when employing combined EF and SQD methods across various active spaces of differing sizes. The results indicate SQD's capability to surpass traditional methods, with energies converging to those obtained by gold-standard coupled-cluster (CCSD) computations for smaller active spaces, while retaining significant accuracy even in larger spaces.
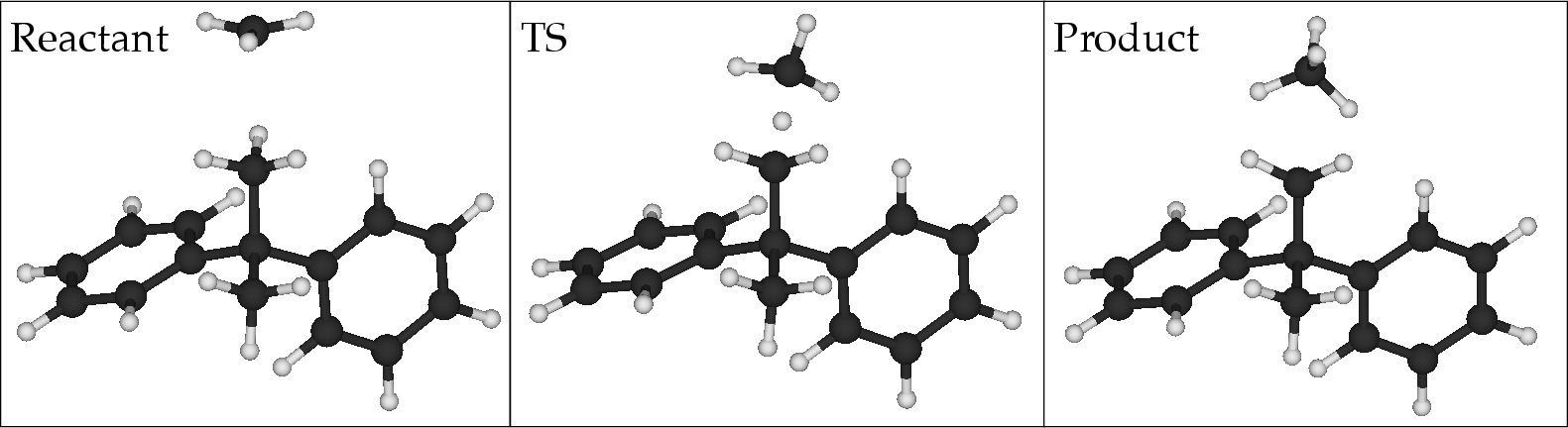
Figure 2: Left to right: reactant, transition state, and product geometries for the hydrogen abstraction reaction considered in this study.
Validation and Comparison
Quantitative validation of the approach using deviations from DMRG benchmark energies revealed the SQD's proficiency (Figures 6 and 7). The simulation's deviation analysis underscores its capability to maintain accuracy within practical limits, highlighting the methodology's efficiency for larger computational problems typical in quantum chemistry.
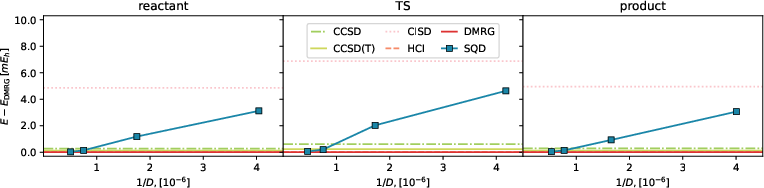
Figure 3: Deviation between SQD and DMRG energy (blue squares) as a function of the subspace dimension for reactant, transition state, and product (left to right) in a (13e,13o) active space, and classical methods (horizontal lines).
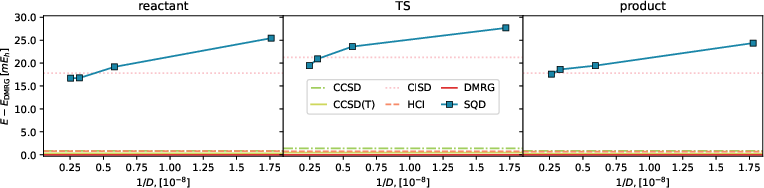
Figure 4: Deviation between SQD and DMRG energy (blue squares) as a function of the subspace dimension for reactant, transition state, and product (left to right) in a (23e,23o) active space, and classical methods (horizontal lines).
Implications and Future Work
The integration of EF with SQD within a quantum-centric simulation framework represents a significant advancement for quantum chemistry, especially for handling reactions of industrial importance. This methodology holds promise for substantial reductions in computational overhead, potentially expediting the material design and analysis processes. Further, the research opens avenues for exploring extended EF functionalities and potential hybrid classical-quantum solutions for complex chemical systems.
Conclusion
This work has successfully combined entanglement forging and sample-based quantum diagonalization into a coherent framework that significantly enhances the simulation of hydrogen abstraction reactions in radical chain processes. This methodological advancement supports more detailed and efficient modeling of degradation processes in materials, presenting promising implications for both applied science and theoretical exploration in quantum computational chemistry.
