Protein Circuit Tracing via Cross-layer Transcoders
Abstract: Protein LLMs (pLMs) have emerged as powerful predictors of protein structure and function. However, the computational circuits underlying their predictions remain poorly understood. Recent mechanistic interpretability methods decompose pLM representations into interpretable features, but they treat each layer independently and thus fail to capture cross-layer computation, limiting their ability to approximate the full model. We introduce ProtoMech, a framework for discovering computational circuits in pLMs using cross-layer transcoders that learn sparse latent representations jointly across layers to capture the model's full computational circuitry. Applied to the pLM ESM2, ProtoMech recovers 82-89% of the original performance on protein family classification and function prediction tasks. ProtoMech then identifies compressed circuits that use <1% of the latent space while retaining up to 79% of model accuracy, revealing correspondence with structural and functional motifs, including binding, signaling, and stability. Steering along these circuits enables high-fitness protein design, surpassing baseline methods in more than 70% of cases. These results establish ProtoMech as a principled framework for protein circuit tracing.
Paper Prompts
Sign up for free to create and run prompts on this paper using GPT-5.
Top Community Prompts
Explain it Like I'm 14
What is this paper about?
This paper introduces a method called ProtoMech that helps us “peek inside” powerful AI models that read protein sequences. These models, called protein LLMs (pLMs), are great at guessing a protein’s structure and what it does, but we don’t really know how they make their decisions. ProtoMech traces the model’s internal “circuits” so we can see which parts of the model are doing what, and then uses that knowledge to design better protein variants.
What questions are the authors trying to answer?
- Can we find the actual step‑by‑step “routes” (circuits) inside a protein model that lead to its predictions?
- Can we compress these routes into a small set of simple, human‑understandable features without losing much accuracy?
- Do these routes match real biology, like known binding sites or important motifs in proteins?
- Can we use these routes to purposefully tweak the model and design higher‑fitness protein variants?
How did they do it? (Explained with simple analogies)
Think of a protein LLM like a many‑layered “reader” that turns a protein’s letters (amino acids) into meaning, similar to how a LLM reads sentences. Inside this reader are many steps (layers). At each step, the model transforms what it knows so far. The tricky part is that the model’s inner workings are super complex and hard to interpret.
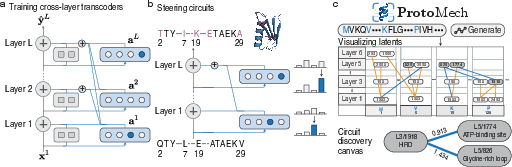
ProtoMech builds a “replacement model” that imitates the original model’s inner steps but makes them easier to understand:
- Layers as steps in a recipe: The original model has multiple layers. ProtoMech recreates what each layer outputs, but in a simpler, more explainable way.
- Latent features as tiny switches: ProtoMech represents information using many small “switches” (features). Only a few turn on at a time—this is called sparsity. You can think of it like choosing the few strongest clues and ignoring the rest.
- TopK as “pick the top few”: At each step, ProtoMech keeps only the top k strongest features and sets the others to zero. It’s like keeping only the loudest signals in a noisy room.
- Cross‑layer transcoders as translators across steps: Most older methods look at each layer separately, like judging a recipe by tasting just one step. ProtoMech’s cross‑layer transcoders connect the features across layers, so a feature turned on early can influence later steps. This captures the full chain of reasoning, not just one moment.
- Replacement model: During testing, ProtoMech swaps in its simple circuits for the original model’s internal parts, so we can see if these circuits can faithfully do the same job.
They tested ProtoMech on two kinds of tasks:
- Protein family classification: Does a protein belong to a certain family?
- Function (fitness) prediction: How good is a protein variant likely to be at its job, based on mutation data?
They also “steered” the model:
- Steering means nudging certain switches on purpose to push the model toward designing better protein variants. Imagine turning specific knobs you know control “binding strength” or “stability.”
Finally, they built a visualizer:
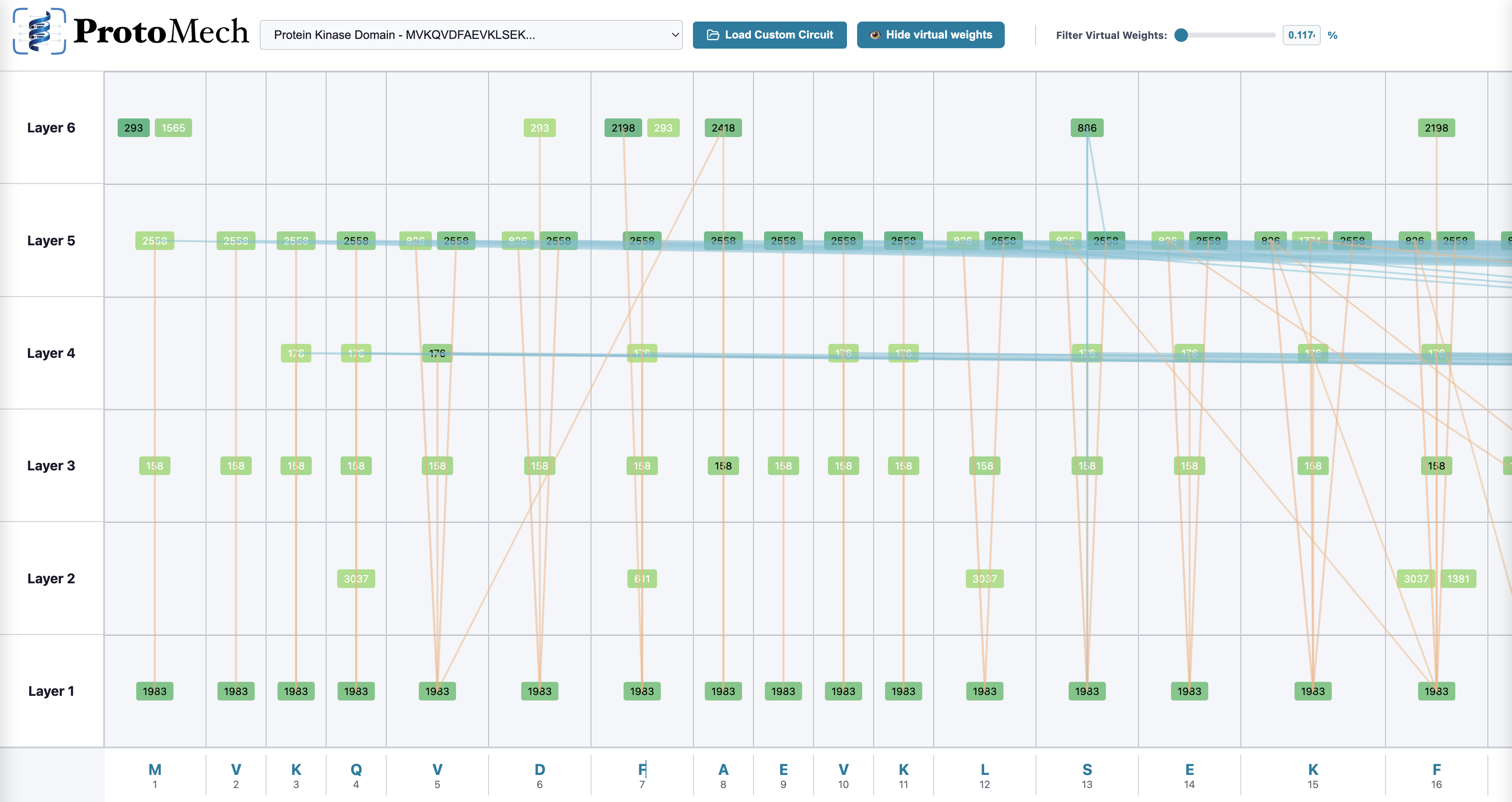
- This tool draws a sparse graph of the most important switches and how they connect across layers, then links them to known biological patterns (motifs) and structures.
What did they find, and why is it important?
Here are the main results in simple terms:
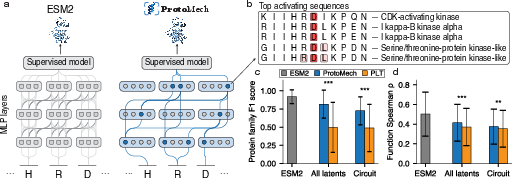
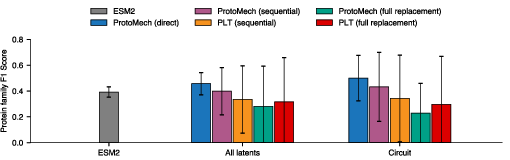
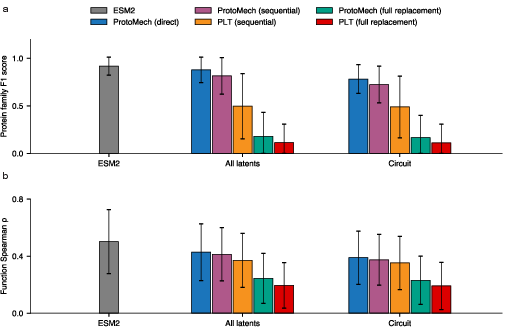
- ProtoMech matches most of the original model’s skill:
- It recovers about 89% of the original accuracy on family classification and 82% on function prediction.
- It compresses the model’s reasoning:
- ProtoMech can keep up to 74–79% of the original performance using less than 1% of the available feature space. That means a tiny set of switches drives most of the useful behavior.
- It beats older methods that ignore cross‑layer reasoning:
- A baseline that treats each layer separately (PLT) performs worse. Cross‑layer connections really matter.
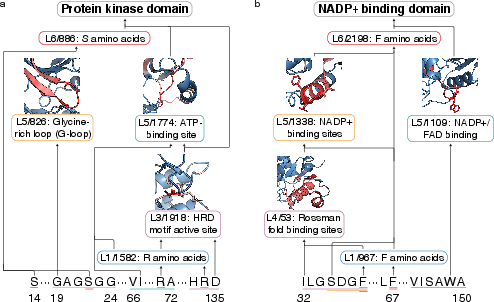
- It discovers real biological motifs:
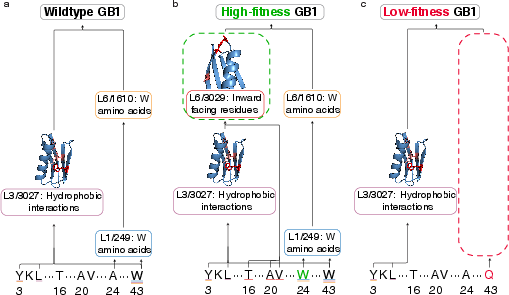
- The circuits align with known patterns, like the HRD catalytic motif in kinases, NADP+ binding pockets, Rossmann folds, and residues important for stability and binding in the GB1 protein.
- It helps design better proteins:
- When steering along the discovered circuits, ProtoMech generated higher‑fitness variants more often than baseline methods, producing the best sequences in over 70% of tests.
Why this matters:
- Understanding: It turns black‑box predictions into understandable, step‑by‑step routes.
- Trust and control: If you know which switches control “binding” or “stability,” you can nudge them and predictably change outcomes.
- Discovery: Matching circuits to known motifs supports real biological insight, and may reveal new ones.
What could this change in the future?
- Faster, smarter protein design: By tracing and steering specific circuits, researchers can focus on variants that use biologically meaningful patterns, speeding up screening and improving success rates.
- More reliable models: ProtoMech’s sparse circuits seem to filter out noise, sometimes even outperforming the original model when tasks are hard. That suggests a path to more robust tools.
- Better interpretability tools: ProtoMech provides open‑source code and a visualizer, helping others explore circuits and link them to structure and function.
- Scaling up: One challenge is that cross‑layer models are larger to train. Future work may reduce the cost so this approach can be applied to bigger protein models.
In short, ProtoMech shows that protein LLMs don’t just guess—they follow traceable routes that line up with real biology. By finding and steering these routes, we can both understand and improve protein design.
Knowledge Gaps
Knowledge gaps, limitations, and open questions
Below is a single, concrete list of what remains missing, uncertain, or unexplored, framed to be actionable for future work:
- Full-model tracing: ProtoMech replaces only MLP blocks and holds attention outputs fixed. It does not recover attention-head circuitry or attention–MLP interactions. Develop and evaluate attention transcoders and joint CLTs that trace the complete transformer computation without relying on ground-truth attention.
- Recursive replacement robustness: Fully recursive replacement (attention + MLP) reportedly degrades performance. Systematically characterize error propagation across depth and design error-control mechanisms (e.g., residual balancing, layer-wise normalization, iterative refinement) to enable faithful end-to-end replacement.
- Scaling to larger pLMs: Results are limited to ESM2-8M (6 layers, d_model=320). Assess feasibility, accuracy, and compute trade-offs for larger ESM2 variants (e.g., 650M, 3B), and other architectures (ProtBert, ProGen), including long-context proteins and multi-domain sequences.
- Parameter efficiency: CLTs incur O(L2) decoder matrices and ~3.5× parameter overhead (28M vs. 8M). Explore low-rank factorization, parameter sharing/tied decoders, block-sparsity, mixture-of-experts, or per-layer routing to reduce memory and training cost while preserving circuit fidelity.
- Hyperparameter sensitivity: No systematic analysis of TopK sparsity (k), latent dimension, auxiliary loss weight (α), and k_aux. Perform ablations to quantify effects on reconstruction, circuit compactness, interpretability, and downstream recovery.
- Identifiability and stability of circuits: Evaluate whether discovered circuits are unique or multiple near-minimal circuits exist. Measure cross-run stability (different seeds, datasets), agreement metrics (Jaccard, overlap), and sensitivity to initialization and training noise.
- Attribution-based selection biases: Circuit discovery uses greedy gradient attribution. Compare against alternatives (Integrated Gradients, DeepLIFT, Shapley approximations, backward elimination, convex feature selection) and quantify robustness, minimality, and false-positive/false-negative rates.
- Probe dependence: Circuits are defined relative to supervised probes (logistic regression for families, CNN for fitness). Test probe invariance by varying probe architectures/capacities and using non-parametric evaluation (e.g., rank-based metrics directly from model logits) to ensure circuits reflect pLM computation rather than probe artifacts.
- Task breadth: Evaluation is limited to family classification and DMS fitness. Extend to structure and contact prediction, binding-site identification, subcellular localization, stability datasets (e.g., Meltome), epistasis modeling, and allosteric effects to test generality of circuit recovery.
- OOD generalization: Assess circuit fidelity on out-of-distribution sequences (de novo designs, disordered proteins, synthetic libraries), across diverse taxa, and multi-domain proteins, including domain mixing and context shifts.
- Dataset biases and confounders: Quantify whether circuits capture trivial features (length, composition, taxonomy) vs. true motifs. Control for sequence identity, family overlap, and dataset clustering (30% identity), and test circuits on carefully confounded/controlled splits.
- Quantitative interpretability metrics: Beyond case studies, introduce systematic measures of motif alignment (precision/recall against Pfam/InterPro), structural enrichment (contact-map overlap, proximity to binding pockets), and coverage across latents and layers.
- Automated circuit annotation: Current motif labeling is manual. Build automated pipelines mapping latents to sequence motifs, secondary structure, 3D contacts (via PDB/AlphaFold), ligand-binding annotations, and GO terms, with confidence scoring and human-in-the-loop curation.
- Layer-wise semantics: The claim that early layers capture residues and later layers capture motifs is qualitative. Statistically validate layer-specific concept types, quantify cross-layer dependencies, and test whether such progression generalizes across tasks and proteins.
- Edge validity in visualizations: Edges are activation × gradient (local linearization). Validate edge causality via targeted ablations (zeroing latents/edges), counterfactual interventions, and measuring effect sizes on probe outputs and reconstruction error.
- Dead latents and utilization: The auxiliary loss aims to reduce inactive units, but latent usage distributions and task-specific specialization are not reported. Quantify active/inactive latents per layer, usage diversity across tasks, and whether dead units persist.
- Denoising claim: Circuits sometimes outperform ESM2 (regularization effect). Formally test denoising by injecting synthetic noise, assessing adversarial robustness, calibration, and uncertainty, and by reusing ProtoMech latents as features for downstream tasks.
- Steering evaluation without wet-lab validation: Fitness gains are judged via a CNN proxy. Validate designed variants experimentally (wet lab) and/or with orthogonal predictors (e.g., structural stability, binding affinity simulations) to confirm true functional improvements.
- Steering baselines: Comparisons include PLT, CAA, and random. Benchmark against stronger design methods (EVE/GEMME, ProT-VAE, structure-guided design, gradient-based sequence optimization), and multi-objective settings (function + stability + solubility).
- Steering methodology details: Activation clamping uses per-node max×scalar heuristic. Conduct sensitivity analysis over scalar choice, node selection strategies (single vs. multi-node), intervention timing (which layers), and constraints (structure-preserving, epistasis-aware optimization).
- Mutational radius constraint: Generation was limited to ≤5 mutations from wildtype. Evaluate steering beyond local neighborhoods, global exploration, and the impact on fitness landscapes with rugged topology and long-range epistasis.
- Attention–MLP interaction during steering: Steering operates through MLP latents and decodes to logits; attention is not jointly controlled. Investigate steering that co-modulates attention heads, and measure synergies or conflicts between attention and MLP interventions.
- Calibration and uncertainty: Report F1 and Spearman but not calibration, confidence intervals across tasks, or uncertainty estimates. Provide calibrated metrics, bootstrap CIs, and per-family/per-assay reliability to inform deployment.
- Reproducibility: While code is released, document seeds, exact hyperparameters, training schedules, compute budgets, and data splits to enable strict replication. Provide pretrained CLTs and standardized benchmarks for circuit discovery/steering.
- Theoretical guarantees: CLTs with TopK sparsity lack identifiability guarantees for recovering “true” circuits. Analyze the conditions under which CLTs faithfully recover mechanistic pathways (e.g., sparsity assumptions, incoherence, mutual information), and derive bounds on reconstruction and attribution reliability.
- Cross-task circuit reuse: It is unclear whether circuits discovered for one task transfer to related tasks (e.g., kinase family classification → kinase activity prediction). Test transferability, modularity, and composability of circuits across tasks and proteins.
- Biological coverage limits: Annotation focuses on well-characterized motifs (kinase, Rossmann fold, GB1). Systematically examine circuits for less-characterized domains/functions to discover novel motifs, and quantify novelty vs. known biology.
- Ethical and safety considerations for design: Steering toward high fitness could inadvertently optimize undesirable properties (e.g., toxicity, immune evasion). Incorporate safety filters (immunogenicity, pathogenicity screens) and report safeguards for responsible use.
Practical Applications
Below is a concise mapping from the paper’s findings to practical, real‑world applications. Items are grouped by deployment horizon and, for each, we outline target sectors, what the application does, how it would be used, tools/workflows that could emerge, and key assumptions/dependencies.
Immediate Applications
- Mechanistic motif discovery and annotation
- Sectors: academia, bioinformatics databases (e.g., UniProt/InterPro), healthcare R&D
- What: Use ProtoMech’s cross‑layer latent circuits and visualizer to reveal and label structural/functional motifs (e.g., kinase HRD motif, Rossmann fold, ATP/NADP+ binding pockets), improving interpretability of protein LLMs and facilitating annotation of uncharacterized proteins.
- How/Workflow: Run ESM2+ProtoMech on protein sets; inspect high‑attribution latents per family; cross‑reference top activating sequences with known motifs; overlay on structures to propose annotations.
- Tools/Products: “ProtoMech Visualizer” (released), plug‑ins for InterPro/UniProt curators, internal motif‑mining dashboards.
- Assumptions/Dependencies: Availability of trained CLT for ESM2-8M (code released); motif labels and structures for validation; interpretability accuracy may vary across families.
- Circuit‑guided candidate prioritization for protein engineering screens
- Sectors: biotech, pharmaceuticals, industrial enzymes, gene therapy (AAV), diagnostics
- What: Use sparse circuits as a mechanistic filter to triage large sequence libraries and enrich for candidates using biologically plausible pathways.
- How/Workflow: Generate or collect variant libraries, compute circuit attributions, filter to variants activating desired functional circuits; send shortlisted variants to wet‑lab screening.
- Tools/Products: “Circuit Filter” module for design‑build‑test pipelines; integration with LIMS/ELN and HTS robotics.
- Assumptions/Dependencies: Empirical validation still required; circuit relevance may be task‑specific; benefits depend on the target’s representation in ESM2.
- Local circuit steering for high‑fitness mutation proposals
- Sectors: biotech/pharma (enzyme optimization, binding domains), gene therapy capsid engineering, synthetic biology
- What: Apply activation clamping on discovered function circuits to propose beneficial substitutions near a wildtype; paper shows superior performance to random and CAA in >70% of tested DMS assays.
- How/Workflow: Select function‑linked circuit nodes, clamp activations to steer residual stream, decode to logits, propose top mutations within an edit radius; validate experimentally.
- Tools/Products: “CircuitMut” API for design teams; Jupyter recipes for GFP, GB1, AAV capsid tasks.
- Assumptions/Dependencies: Demonstrated strongest within small mutation radii (≤5); relies on ESM2+CLT pairing and DMS‑like regimes; wet‑lab confirmation needed.
- Focused combinatorial library design for directed evolution
- Sectors: industrial biocatalysis, therapeutic enzymes, materials enzymes, agriculture
- What: Use circuits that highlight “gatekeeper” residues and interacting motifs to define positions for saturation mutagenesis or combinatorial libraries.
- How/Workflow: Identify residues supported by cross‑layer circuits (e.g., binding/stability hotspots), design concise libraries centered on these positions, increase hit rate per screened variant.
- Tools/Products: “Circuit‑guided library designer” that exports oligo pools or mutagenesis plans.
- Assumptions/Dependencies: Requires accurate mapping from latents to residues; structural context helps; still necessitates throughput capacity and assay design.
- Model debugging and interpretability audits for pLMs
- Sectors: software/AI for biology, MLOps, quality assurance in regulated R&D
- What: Use replacement model and circuits to audit why a pLM succeeds/fails on tasks; identify spurious features, denoise representations (paper notes circuits sometimes outperform full ESM2).
- How/Workflow: Compare task performance with full vs. circuit‑compressed paths; examine attribution graphs to flag brittle features; guide dataset augmentation or model retraining.
- Tools/Products: “Circuit Audit Dashboard” for pLM interpretability, drift monitors using circuit activation distributions.
- Assumptions/Dependencies: Requires engineering effort to integrate with existing pLMs; interpretability conclusions need domain review.
- Hypothesis generation and experimental design in molecular biology courses and labs
- Sectors: education, academic research
- What: Teach structure‑function relationships with interactive circuit graphs; design mutation experiments to test circuit‑predicted motifs.
- How/Workflow: Load sequences, inspect circuits, plan point mutations targeting latent‑identified motifs; compare predictions vs. assay outcomes.
- Tools/Products: Courseware using the ProtoMech visualizer; structured lab modules.
- Assumptions/Dependencies: Access to basic computational infrastructure; simplified tasks advisable for instruction.
- Functional annotation assistance for uncharacterized proteins
- Sectors: bioinformatics, microbiome/novel protein discovery
- What: Use circuits to flag likely domains, binding pockets, or catalytic residues to prioritize proteins for deeper study.
- How/Workflow: Batch‑process uncharacterized proteins, extract top circuits, cluster by circuit signature, propose function candidates to curators or experimentalists.
- Tools/Products: Batch annotation pipelines; triage reports for curation teams.
- Assumptions/Dependencies: Annotations are putative; best used to prioritize follow‑up rather than as definitive labels.
- Preliminary biosecurity and synthesis screening aid
- Sectors: policy, gene synthesis providers, biodefense
- What: Flag sequences that strongly activate circuits associated with sensitive functions (e.g., specific binding or enzymatic motifs) as an additional review signal.
- How/Workflow: Run submissions through ProtoMech circuits; add risk tags when sensitive motifs are detected; escalate for human review.
- Tools/Products: “Circuit Risk Tagger” augmenting existing sequence screening (e.g., best‑practice frameworks).
- Assumptions/Dependencies: Not a standalone decision tool; false positives/negatives possible; requires governance and expert oversight.
- Task‑specific model compression for inference savings
- Sectors: software tooling for biology R&D, cloud providers, embedded analytics
- What: Replace full MLP pathways with compact circuit subsets that retain 74–79% performance using <1% latents for specific tasks (family classification, some function prediction).
- How/Workflow: Deploy circuit‑restricted inference for high‑throughput triage; fall back to full model for borderline cases.
- Tools/Products: “CircuitProbe” lightweight inference mode; edge‑compatible triage microservices.
- Assumptions/Dependencies: Accepts reduced accuracy; task‑by‑task validation required; attention pathways remain fixed in replacement approach as per paper.
Long‑Term Applications
- Scaling to larger pLMs for complex objectives
- Sectors: pharma (antibodies, enzymes), advanced gene therapy, agriculture, materials
- What: Train CLTs for larger ESM variants or next‑gen pLMs to uncover richer circuits for multi‑domain proteins and complex traits.
- How/Workflow: Optimize CLT training (reduce O(L²) decoder scaling), exploit parameter sharing or low‑rank decoders; retrain on larger corpora.
- Tools/Products: “ProtoMech‑XL” with efficient cross‑layer transcoders.
- Assumptions/Dependencies: Significant compute and engineering; careful regularization to avoid error accumulation.
- Multi‑objective circuit composition for de novo design
- Sectors: therapeutics, industrial enzymes, diagnostics
- What: Compose circuits for stability, binding specificity, solubility, and immunogenicity to steer jointly for multiple design targets.
- How/Workflow: Discover per‑objective circuits; develop weighting/constraint schemes for activation clamping; close the loop with multi‑assay validation.
- Tools/Products: “CircuitComposer” multi‑objective design suite; integration with ProteinMPNN/RFdiffusion for structure compliance.
- Assumptions/Dependencies: Requires robust circuits for each objective and conflict resolution strategies; extensive experimental validation.
- Automated circuit annotation pipelines
- Sectors: academia, databases, tool vendors
- What: Use motif libraries, structural predictions, and literature mining to auto‑label latent circuits at scale.
- How/Workflow: Align circuit‑activated residues to predicted structures; match to motif databases (e.g., PROSITE/InterPro); rank confidence and push to curators.
- Tools/Products: “AutoAnnotator” service; confidence‑scored circuit catalogs.
- Assumptions/Dependencies: High‑quality structure prediction and curated motif ontologies; handling novel/unlabeled circuits remains challenging.
- Closed‑loop autonomous DBTL with circuit‑aware controllers
- Sectors: lab automation, robotics in biotech
- What: Integrate circuit steering with robotic build‑test cycles to rapidly explore productive regions of fitness landscapes.
- How/Workflow: Plan circuit‑guided libraries, execute on liquid handlers, measure fitness, update steering weights/circuits iteratively.
- Tools/Products: Circuit‑aware experiment planners, OT‑2/automation integrations, Bayesian optimization using circuit features.
- Assumptions/Dependencies: Robust assays, integration engineering, informatics infrastructure.
- Regulatory‑grade explainability for AI‑designed biologics
- Sectors: policy/regulatory affairs, pharma QA
- What: Provide mechanistic evidence linking design decisions to interpretable circuits to support regulatory submissions and internal governance.
- How/Workflow: Generate circuit reports for candidate therapeutics showing alignment to known motifs and risk mitigations; track provenance through design iterations.
- Tools/Products: “Mechanism Report” generator for dossiers; audit trails tying circuits to decisions.
- Assumptions/Dependencies: Emerging standards for explainability in drug development; regulators’ acceptance of such evidence will evolve.
- Model monitoring and drift detection via circuit fingerprints
- Sectors: MLOps for bio R&D
- What: Use changes in circuit activation patterns as indicators of input distribution shifts or model drift in production pipelines.
- How/Workflow: Track activation statistics over time; alert when key circuits deactivate or new circuits dominate; trigger retraining or review.
- Tools/Products: Circuit telemetry and alerting; SLA dashboards for pLM‑based services.
- Assumptions/Dependencies: Requires baseline fingerprints and robust thresholds; interpretability team support.
- Cross‑modality extension to DNA/RNA LLMs
- Sectors: functional genomics, gene regulation, mRNA therapeutics
- What: Adapt cross‑layer transcoders to interpret circuits governing promoters, enhancers, splicing, or RNA structure/function.
- How/Workflow: Train CLTs on genomics/trascriptomics LMs; discover circuits for regulatory grammar; steer for expression or splicing outcomes.
- Tools/Products: “GeneProtoMech” and “RNAMech” toolkits.
- Assumptions/Dependencies: Data heterogeneity and context length; need modality‑specific probes and assays.
- Generative control knobs for novel fold/function discovery
- Sectors: advanced therapeutics, materials science, bioenergy
- What: Use circuits as interpretable control knobs in generative pipelines to bias toward novel folds with targeted properties (e.g., cellulase efficiency, CO₂‑fixing enzymes).
- How/Workflow: Couple circuit steering with structure‑conditioned models; enforce fold constraints while steering for catalytic motifs.
- Tools/Products: Circuit‑guided generative co‑design platforms.
- Assumptions/Dependencies: Requires robust structure‑function coupling; extensive wet‑lab validation; higher risk of off‑target effects.
- R&D portfolio and IP strategy support
- Sectors: biotech finance, corporate strategy, IP
- What: Use circuits to de‑risk programs (evidence of mechanism), identify white‑space motifs, and support claim drafting around mechanistic features.
- How/Workflow: Map pipeline assets’ circuits against known biology; assess novelty and breadth; inform go/no‑go and claim scope.
- Tools/Products: Circuit‑based diligence reports.
- Assumptions/Dependencies: Not a legal substitute; requires expert interpretation; depends on model coverage of the target biology.
Notes on global dependencies and feasibility
- Data/compute: Training CLTs used 5M UniRef50 sequences and increases parameters (~3.5× for ESM2‑8M). Scaling up requires additional optimization.
- Replacement model design: Paper’s best results fix attention outputs and replace MLPs; fully recursive replacements degrade performance—this should guide deployment choices.
- Generalization: Strongest evidence is on specific families/assays (e.g., kinases, NADP+, GB1, GFP, AAV). New domains may need task‑specific evaluation.
- Experimental validation: Steering and prioritization produce hypotheses; wet‑lab confirmation is essential, especially for therapeutic or safety‑critical use.
- Interpretability limits: Some circuits may reflect unknown biology; automated annotation is nascent and should be used to prioritize rather than conclusively label.
Glossary
- Activation clamping: Forcing selected latent units to fixed high values during a forward pass to steer model behavior. "Our steering strategy involves activation clamping within the CLT replacement model"
- Attribution score: A gradient-based measure of how much a latent unit contributes to a probe’s output. "we compute an attribution score by measuring its contribution to the probe’s output on a held-out validation set"
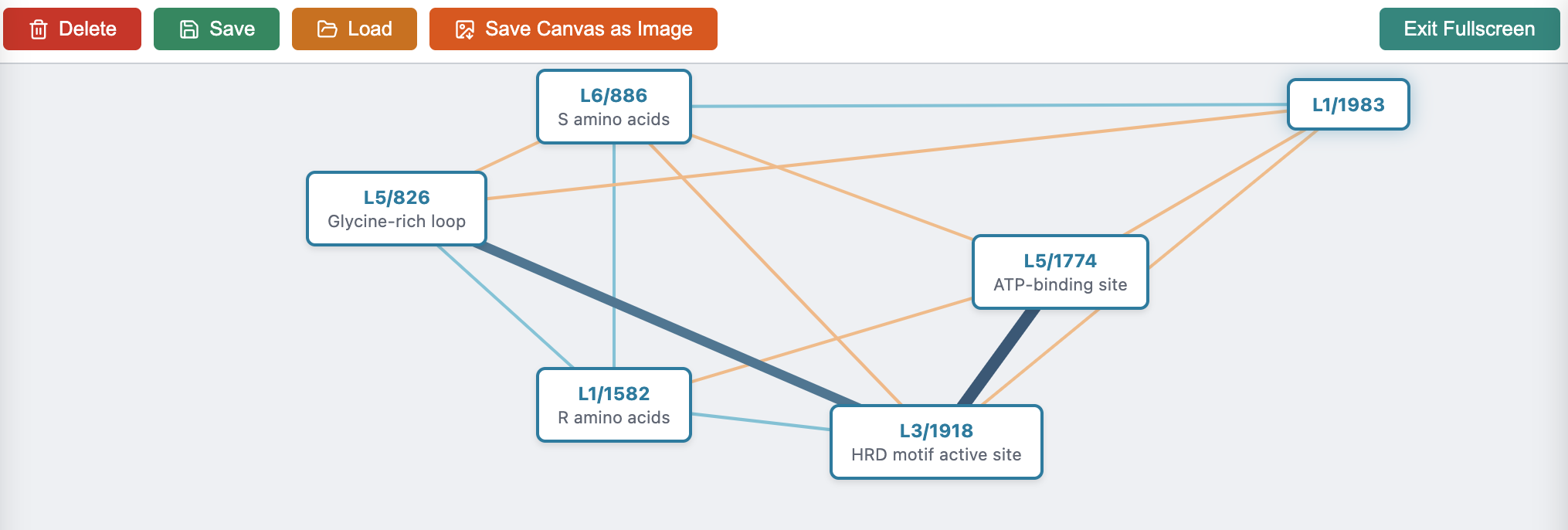
- ATP-binding site: A protein pocket where ATP docks to enable phosphorylation. "L5/1774 identifies the ATP-binding site"
- Auxiliary loss: An additional training objective used to encourage active latents and improve reconstructions. "we incorporate an auxiliary loss"
- Concept vector: A direction in activation space representing a target concept, often estimated by mean differences between groups. "a global ``concept vector" in ESM2's hidden dimension"
- Contrastive Activation Addition (CAA): A steering method that injects a concept vector into hidden activations to bias outputs. "Contrastive Activation Addition (CAA)"
- Convolutional neural network (CNN) probe: A supervised model trained on internal representations to predict fitness or labels. "we train a convolutional neural network (CNN) probe"
- Cross-layer transcoders (CLTs): Replacement models that reconstruct each layer’s outputs from sparse latents across all preceding layers. "CLTs extend standard transcoders"
- Dead latent units: Latent features that remain inactive during training and contribute nothing to reconstructions. "inactive (“dead”) latent units"
- Decoder matrix: A linear mapping from latent features to reconstructed layer outputs. "a decoder matrix mapping latent features"
- Deep Mutational Scanning (DMS): High-throughput assays measuring the effects of many mutations on protein fitness. "Deep Mutational Scanning (DMS) assays"
- Edge weight: A quantity defining connection strength in attribution graphs, often computed as activation times a gradient. "we compute the edge weight connecting a source node to a target node"
- Encoder matrix: A linear mapping from residual activations to latent features. "the encoder matrix"
- ESM2-8M model: A small variant of the ESM2 protein LLM with six layers and 320-dimensional hidden states. "We train CLTs on the ESM2-8M model"
- Fitness landscapes: Mappings from sequence variants to measured or predicted fitness values. "protein fitness landscapes"
- Gradient-based attribution: A technique that uses gradients to estimate how much components (e.g., latents) influence an output. "based on gradient-based attribution"
- Glycine-rich loop (G-loop): A conserved kinase motif that helps anchor ATP during catalysis. "Glycine-rich loop (G-loop)"
- HRD motif: A conserved catalytic triad in kinases central to phosphorylation. "the HRD motif—a highly conserved catalytic loop"
- Hydrophobic core: The interior of a protein where nonpolar residues cluster, contributing to stability. "buried in GB1's hydrophobic core"
- Hydrophobic interactions: Nonpolar contacts between residues that stabilize protein structure. "which detects hydrophobic interactions"
- InterPro: A database that integrates protein family and domain annotations. "InterPro family labels"
- Latent bottleneck: A restricted set of latent units that forces compact, interpretable representations of activations. "passing activations through a sparse latent bottleneck"
- Latent space: The vector space of latent features representing compressed model information. "Sparsity in the latent space is enforced"
- Latent variables: Individual units in the latent space that capture interpretable features. "subset of latent variables"
- Logistic regression probe: A linear classifier used to evaluate how well internal representations support a task. "we train a logistic regression probe"
- Mechanistic interpretability: Understanding model internals by identifying and analyzing the circuits and features that drive behavior. "Recent progress in mechanistic interpretability"
- Mean-squared error: A reconstruction loss defined as the average squared difference between predicted and target activations. "minimizing the mean-squared error"
- MLP block: The feed-forward sublayer within a transformer layer. "the MLP block"
- NADP+ (Nicotinamide Adenine Dinucleotide Phosphate): A redox cofactor whose binding sites are recognized in certain protein domains. "Nicotinamide Adenine Dinucleotide Phosphate (NADP+)"
- Per-layer transcoders (PLTs): Layer-local transcoders that approximate each layer independently using sparse latents. "per-layer transcoders (PLTs)"
- Protein LLMs (pLMs): Transformer models trained on protein sequences to learn structure- and function-relevant representations. "Protein LLMs (pLMs)"
- ProteinGym: A benchmark of DMS datasets and evaluation protocols for protein fitness prediction. "ProteinGym"
- Replacement model: A surrogate model that emulates a network’s internal computations to enable circuit tracing. "serves as a replacement model for ESM2"
- Residual stream activation: The activation vector in a transformer’s residual pathway before the MLP block. "the residual stream activation prior to the MLP block"
- Rossmann fold: A nucleotide-binding structural motif composed of alternating beta strands and alpha helices. "the Rossmann fold"
- Sparse autoencoders (SAEs): Autoencoders with sparsity constraints that decompose activations into interpretable features. "sparse autoencoders (SAEs)"
- Spearman rank correlation: A nonparametric correlation metric used to assess fitness prediction quality. "the Spearman rank correlation"
- Steering: Intentionally modifying internal activations or features to drive desired outputs. "Steering along these circuits enables high-fitness protein design"
- Swiss-Prot: A curated protein sequence database used for tasks like family classification and motif analysis. "Swiss-Prot"
- TopK activation function: An operator that keeps only the k largest-magnitude activations and zeroes the rest to enforce sparsity. "TopK activation function"
- Transcoders: Modules that approximate transformer MLP mappings via sparse latent representations. "using transcoders"
- UniRef50: A redundancy-reduced protein sequence collection clustered at 50% identity. "UniRef50"
Collections
Sign up for free to add this paper to one or more collections.