- The paper demonstrates that TDΔSCF mitigates functional sensitivity in TDDFT by using a non-Aufbau reference to accurately model near-degenerate states.
- It employs a linear-response framework retaining the full DFT kernel, yielding smoother torsional curves and reliable singlet–triplet energy gaps in diradicals.
- Numerical tests on ethylene, benzyne, and bond dissociation benchmark cases validate the method’s performance despite challenges like orbital instability and overestimated singlet energies.
TDΔSCF: Linear-Response TDDFT with Non-Aufbau References for Near-Degenerate States
Introduction
The paper "TDΔSCF: Time-Dependent Density Functional Theory with a Non-Aufbau Reference for near-degenerate states" (2603.29948) presents a rigorous investigation into a novel linear-response protocol—TDΔSCF: Time-Dependent Density Functional Theory with a non-Aufbau self-consistent field reference—targeting electronic structure problems where near-degeneracy or static correlation severely impedes the reliability of conventional single-reference DFT/TDDFT methodologies. The formalism is compared extensively with standard collinear spin-flip TDDFT (SF-TDDFT), with a focus on fundamental differences in kernel structure, functional dependence, the quality of predicted potential energy surfaces (PES), evaluation of singlet–triplet (S–T) gaps, molecular structure optimization, bond dissociation, and the exposure of an intrinsic numerical instability when using non-Aufbau determinants.
TDΔSCF is constructed by taking as its reference determinant a variationally optimized non-Aufbau (open-shell) solution obtained via constrained SCF (e.g., MOM, STEP), and performing spin-conserving (single excitation) linear-response TDDFT in the Tamm–Dancoff approximation (TDA). In contrast, collinear SF-TDDFT employs a high-spin reference (typically a triplet MS=1), with the target (low-spin) states accessed via spin-flip excitation operators; here, the Coulomb and standard XC kernel contributions vanish in the effective coupling—a result which leaves the SF protocol highly sensitive to the admixture of Hartree–Fock exchange and yields pronounced functional dependence and stability problems, especially for pure and semi-local functionals.
In TDΔSCF, the response manifold is built on the promoted, often open-shell, SCF determinant (Figure 1), so that for near-degenerate configurations (e.g., biradicals, bond-breaking, twisted π systems), both the static correlation and the dynamical response from conventional hybrid or (meta-)GGA functionals remain present in the excitation kernel.
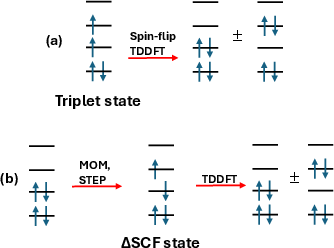
Figure 1: Schematic comparison of SF-TDDFT and TDΔSCF reference choices and excitation spaces.
Ethylene Torsion Benchmark
Torsion about the C=C bond in ethylene is a canonical example of a system with changing multiconfigurational character. Standard DFT/TDDFT curves show unphysical cusps near 90°, indicative of the breakdown of the single-reference ansatz. TDΔSCF yields a smooth, physically plausible torsional PES for all three functionals (BLYP, B3LYP, BHHLYP), closely reproducing the MRCISD high-level reference near the degeneracy point without the abrupt non-differentiability seen in SF-TDDFT (Figure 2), especially for pure functionals.
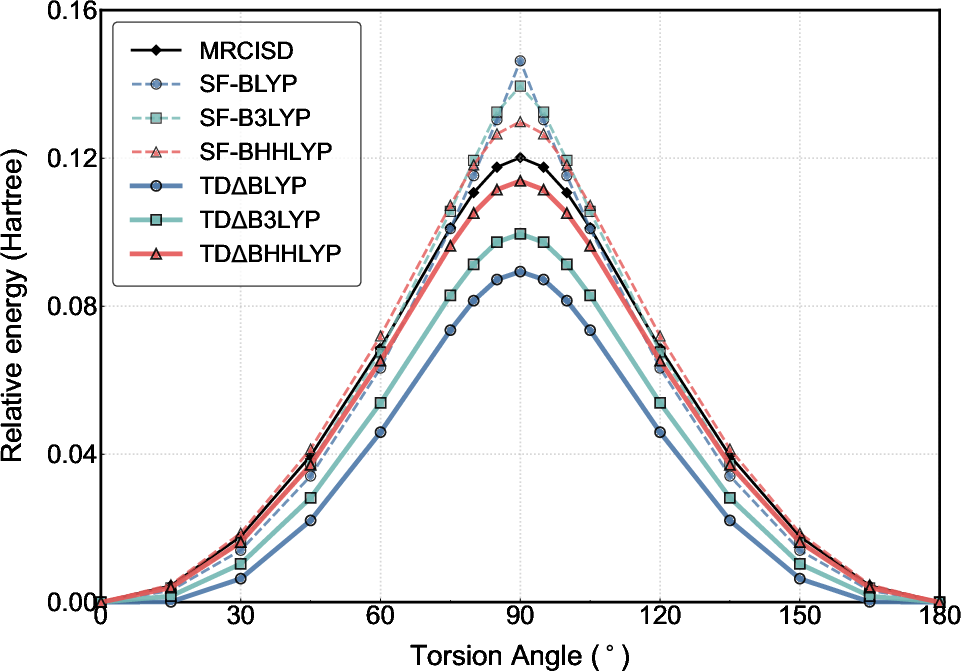
Figure 2: Ethylene torsional barriers calculated using TDΔSCF and SF-TDDFT. TDΔSCF gives smooth barriers, free of cusps present in SF-BLYP.
While SF-TDDFT dramatically overestimates the torsional barrier with pure functionals, TDΔ0SCF exhibits much weaker functional sensitivity, underestimating barriers primarily due to deterioration of the Δ1 end-point—an area still adequately described by ground-state DFT. Re-evaluating barrier heights with KS-DFT as reference for the planar geometry restores quantitative agreement across functionals, revealing that TDΔ2SCF's deficiencies are localized to the non-degenerate regime, consistent with its design focus.
Singlet–Triplet Gaps in Diradicals and Benzyne Isomers
S–T gaps for a set of atomic and molecular diradicals are systematically evaluated. Conventional DFT struggles due to single-determinant bias, with systematic errors growing with HF exchange fraction. SF-TDDFT, especially with BLYP, fails catastrophically (MAE > 200 kcal/mol); the error decreases with hybrid functionals but remains functional-dependent and biased toward underestimating gaps, especially for atoms and one-center diradicals. In contrast, TDΔ3SCF consistently produces accurate gaps with a weak dependence on the underlying functional (MAEs: 4.3–4.7 kcal/mol for BLYP/B3LYP/BHHLYP; Figure 3), always avoiding the catastrophic failures of SF-TDDFT.
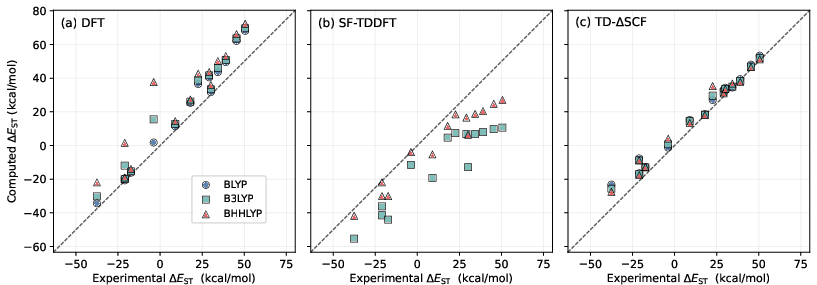
Figure 3: Comparison of computed and experimental singlet–triplet energy gaps (Δ4) for a benchmark set, highlighting the strong functional dependence and severe errors of SF-TDDFT (BLYP) and the modest, weakly functional-dependent errors of TDΔ5SCF.
Importantly, TDΔ6SCF does systematically overestimate S–T gaps compared to experiment, reflecting a bias inherited from the non-Aufbau nature of the reference and lack of optimization on the singlet surface in the response step.
Optimization of Benzyne Isomer Singlet Geometries
Geometry optimizations on o-, m-, and p-benzyne singlet surfaces further reveal functional and methodological differences. For o- and p-benzyne, both protocols yield comparable, reliable geometries (deviation <0.04 Å, <6° from SF-CCSDT reference). For m-benzyne, SF-TDDFT with B3LYP predicts an unphysical bicyclic structure, consistent with known functional pathologies; TDΔ7SCF, by contrast, retains the monocyclic minimum preferred by high-level benchmark and quantum Monte Carlo calculations, with systematic agreement among various functionals (Figure 4).
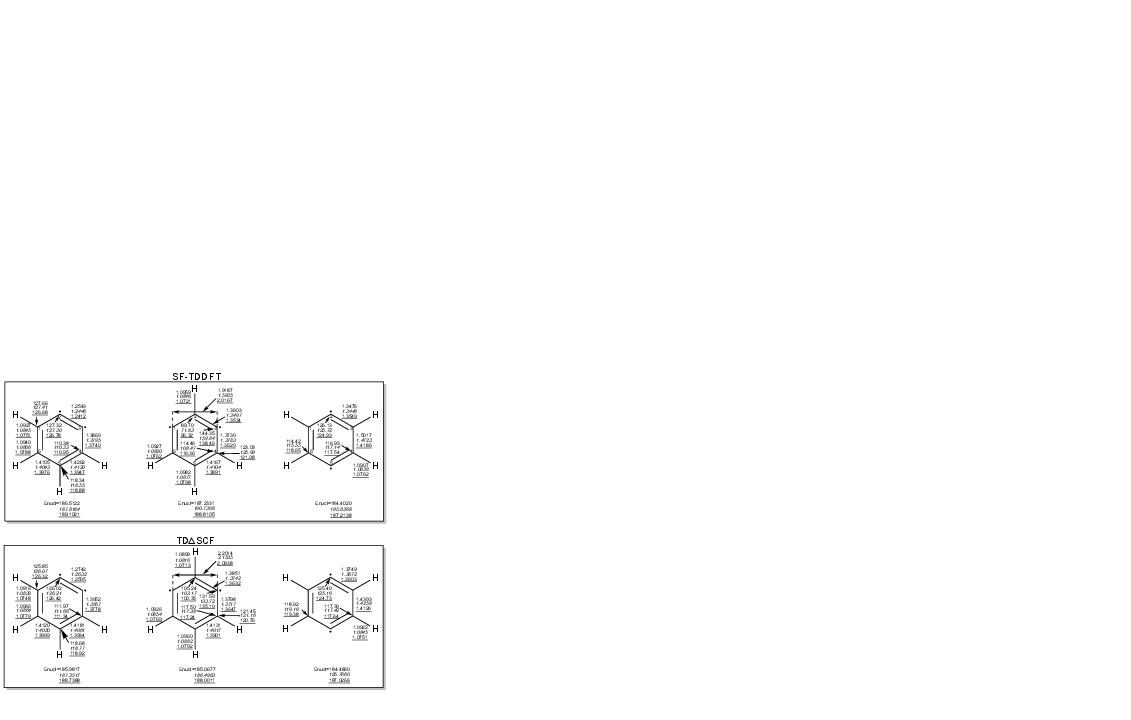
Figure 4: Singlet-state optimized geometrical parameters of benzyne isomers. TDΔ8SCF predicts a monocyclic minimum for singlet m-benzyne for all functionals, unlike SF-B3LYP, which stabilizes an erroneous bicyclic structure.
Bond Dissociation: HF and F₂
The ability to recover the correct dissociation limit is a stringent test of a linear-response formalism. In restricted DFT, the dissociation limit is never achieved due to failure to describe correct near-degeneracy and static correlation. SF-TDDFT with conventional functionals produces strong functional dependence, unphysical ionic solutions (e.g., in HF), and spurious low-lying states (e.g., in F₂). It achieves proper dissociation only with high HF-exchange content and only for some bonds. TDΔ9SCF, in stark contrast, approaches the correct singlet-triplet degeneracy limit smoothly and without generating spurious roots, for all tested functionals (Figures 5 and 6). This robustness is achieved because the response is computed on an appropriate broken-symmetry reference.
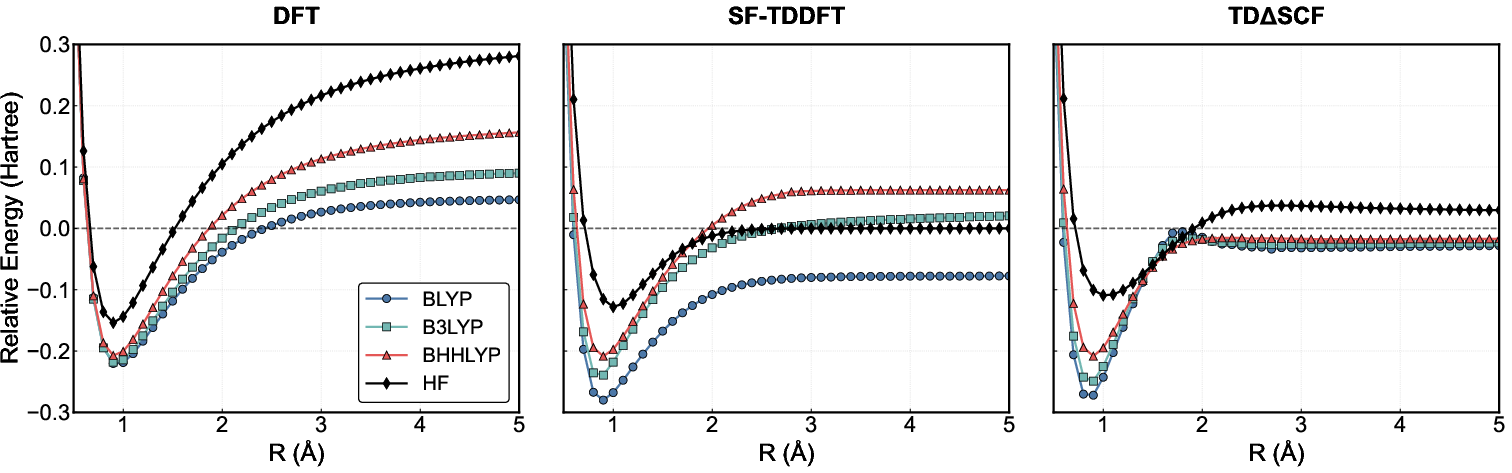
Figure 5: Bond dissociation PES for hydrogen fluoride. TDΔ0SCF (all functionals) properly approaches the unrestricted dissociation limit, free from the ionic root instability present in SF-TDDFT (BLYP/B3LYP).
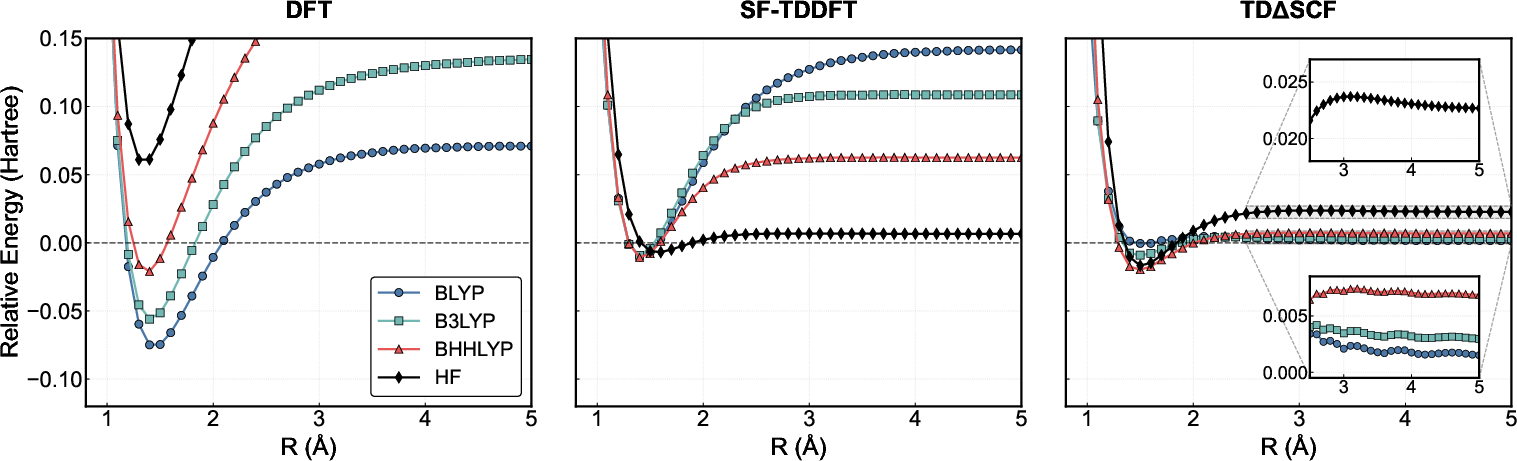
Figure 6: Bond dissociation PES for F₂. Only TDΔ1SCF (all functionals) shows physically reasonable asymptotic behavior without contamination by spurious low-lying roots as seen in SF-TDDFT.
Identification of a Numerical Instability in Non-Aufbau SCF Excitations
A key finding is the first identification and mechanistic analysis of a numerical instability in non-Aufbau SCF and TDΔ2SCF states—arising from divergence in the GGA exchange-correlation potential at points where the reduced gradient Δ3 becomes large on nodal manifolds of singly-occupied antibonding orbitals. In H₂ with a pure Δ4 open-shell configuration, the orbital energies of the antibonding orbital display sharp, grid-dependent discontinuities linked to near-zero density and finite density gradient at nodal points (Figures 7–9). In practical systems, this scenario is rare, but the effect is intrinsic, not merely a numerical artifact, and could be exacerbated in large or strongly correlated systems with pronounced unpaired character at particular spatial regions.
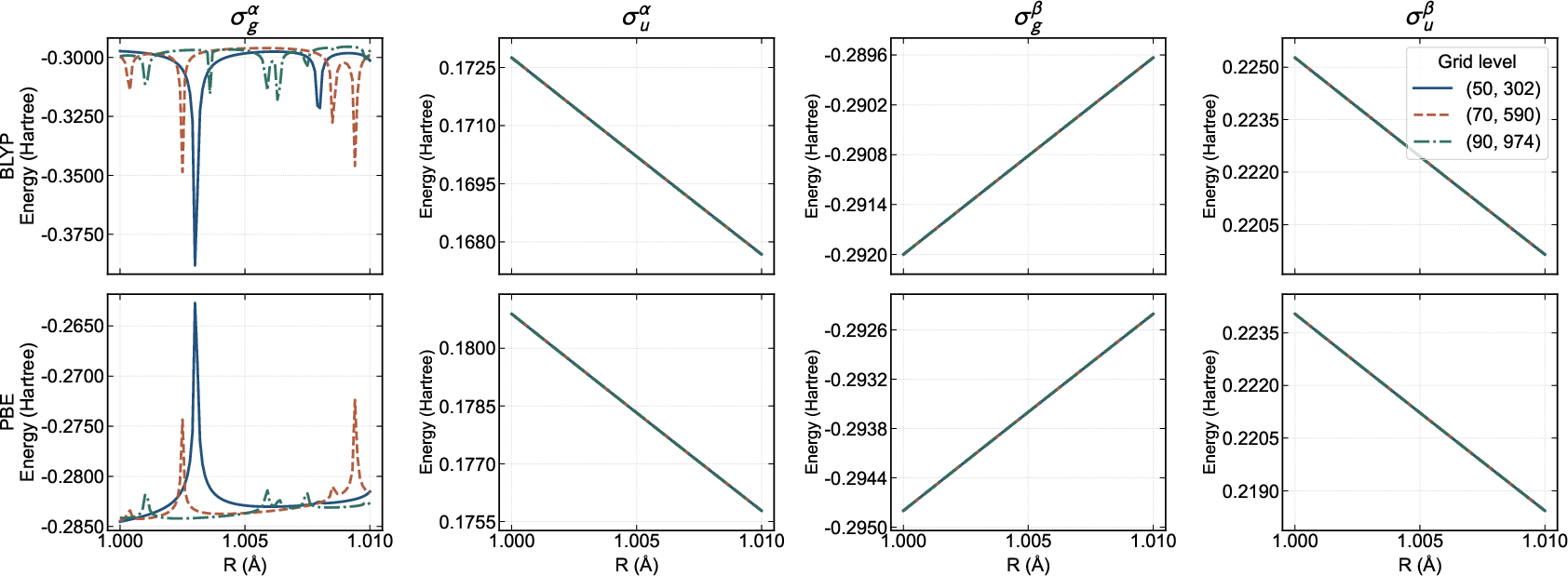
Figure 7: Non-Aufbau SCF orbital energies for H₂, showing sharp peaks in antibonding orbital energy, a direct manifestation of the Δ5 grid instability.
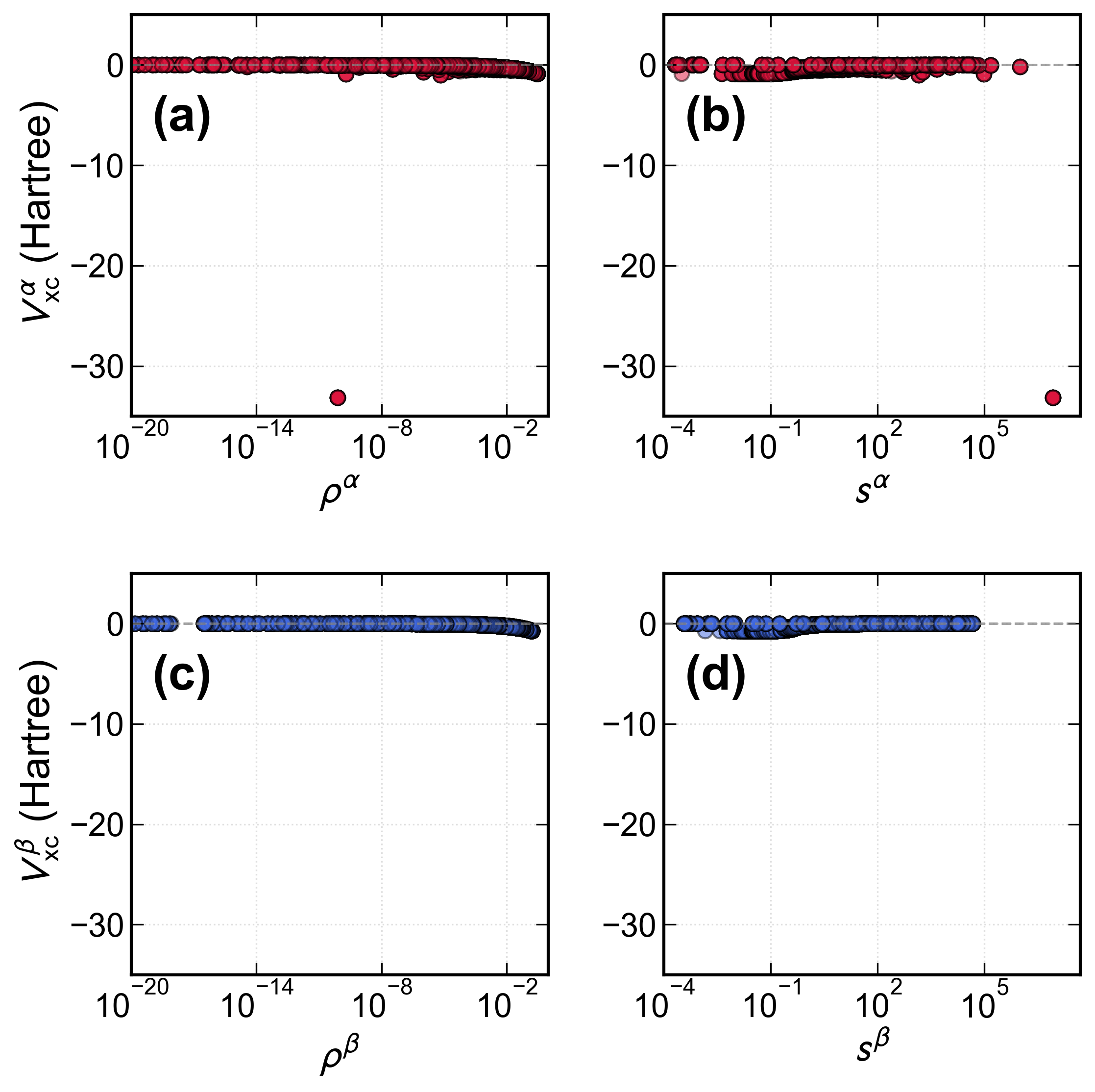
Figure 8: Scatter plots of Δ6 showing divergence at specific grid points with vanishing density and large Δ7.
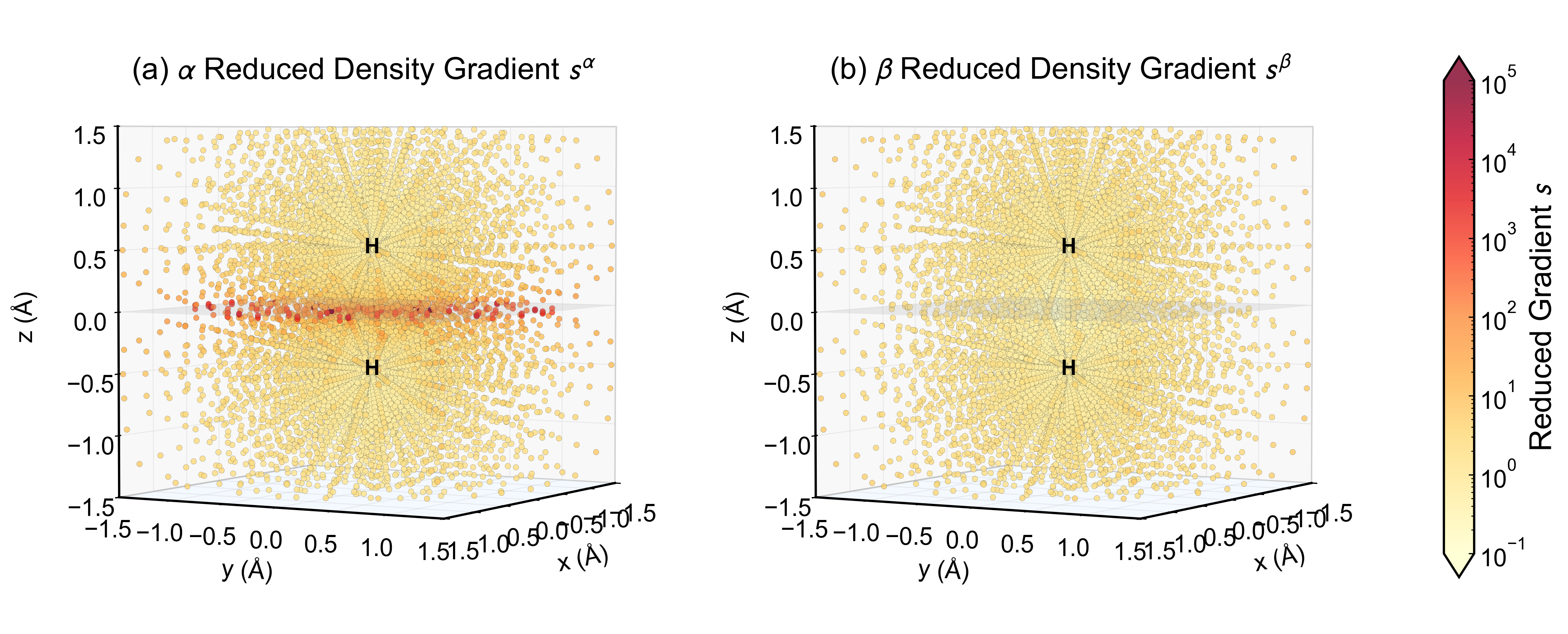
Figure 9: Real-space visualization of Δ8 for H₂; pathology (diverging gradient) is localized at the antibonding nodal plane.
Implications, Limitations, and Theoretical Perspective
TDΔ9SCF establishes itself as a robust, low-cost formalism for challenging single-reference-inadequate problems, offering clearly superior functional insensitivity compared to collinear SF-TDDFT, as well as improved qualitative description for torsion, S–T gaps (especially in "same-center" biradicals), and bond dissociation. Its limitations are noteworthy: the systematic overestimation of singlet energies (hence S–T gaps), performance susceptible to the quality of the underlying non-Aufbau SCF reference, and potential for physically relevant, though rarely encountered, numerical instability when the open-shell density is highly nodal and not compensated by other electrons.
By not requiring explicit construction of noncollinear kernels or the treatment of spin-flip operators, TDΔ0SCF remains straightforward to implement in existing DFT/TDDFT architectures provided robust Δ1SCF optimization protocols (e.g., MOM/STEP/SGM) are available. This also implies that future developments—including automatic diagnosis of the quality of non-Aufbau references, regularization of Δ2 near nodal manifolds, and extension to analytic properties and nonadiabatic couplings—are feasible with minimal infrastructural changes. The approach complements ongoing efforts to generalize TDDFT linear-response or EOM-CC paradigms for strongly correlated and open-shell systems.
Conclusion
TDΔ3SCF defines a new linear-response framework for static correlation and near-degeneracy regimes, eliminating the unphysical functional dependence of SF-TDDFT and delivering accurate PESs, spectroscopic properties, and molecular geometries for problems where single-reference DFT fails. The method exposes and clarifies an intrinsic instability in open-shell DFT for extreme cases, an insight with ramifications for broader developments in density-based excited-state methods. With improvements in reference-state algorithms and regularization strategies for the exchange-correlation potential, TDΔ4SCF is poised for widespread applicability in quantum chemistry and materials science for the description of bond-breaking, conical intersections, and electronically nontrivial molecules.

