- The paper introduces AQVolt26, a comprehensive r2SCAN halide dataset of 322,656 calculations to improve ML potential predictions in solid-state battery modeling.
- It employs high-temperature MD sampling, 2DIRECT clustering, and rigorous DFT labeling to capture complex off-equilibrium ionic transport dynamics.
- The study demonstrates that MLIPs trained with AQVolt26 exhibit superior dynamic stability and ionic conductivity prediction, closely matching experimental benchmarks.
AQVolt26: High-Temperature r2SCAN Halide Dataset for Universal ML Potentials and Solid-State Batteries
Motivation and Context
The urgent demand for safer, high-energy-density electrochemical storage, especially in electric vehicles, has steered attention toward all-solid-state batteries (ASSBs) due to their enhanced safety profiles and improved volumetric energy density. Halide solid-state electrolytes (SSEs) have surfaced as promising candidates for their favorable ionic mobility, electrochemical stability, and mechanical deformability. However, discovering and screening new halide-based SSEs at scale critically depends on the ability of atomistic simulations—particularly molecular dynamics (MD) at elevated temperatures—to reliably sample ion transport pathways across highly anharmonic potential energy surfaces (PES).
Classical force fields, though efficient, lack the transferability and fidelity required to capture the subtle chemical and structural complexities of halide systems. Machine learning interatomic potentials (MLIPs), especially those leveraging large-scale, diverse training datasets, offer a promising alternative if they can be trained to produce robust predictions beyond the near-equilibrium regime. The AQVolt26 study specifically targets the data and modeling gaps in general-purpose MLIPs for dynamically soft halide chemistries, which have flat, shallow PES and facilitate significant ionic excursions under realistic battery conditions.
Dataset Construction and Scope
AQVolt26 introduces a curated dataset of 322,656 r2SCAN-level single-point calculations for Li-containing halide materials, making it the largest off-equilibrium computational resource for solid-state electrolyte modeling. The dataset was assembled through a multi-stage workflow:
- Targeted Compound Selection: 4,911 Li halide candidates, derived from r2SCAN MatPES, the Materials Project, and novel generated structures excluding radioactive, oxidative, or chemically inert elements.
- High-Temperature MD Sampling: Over 200 million configurations were sampled via NpT MD at temperatures up to 1500 K using ML potentials pre-trained on MatPES and MP-ALOE, ensuring coverage of the volatile configurational landscape relevant for superionic transport.
- 2DIRECT Clustering: Configurational diversity was maximized using a two-stage principal component encoding and BIRCH clustering strategy (2DIRECT), ensuring that only 2% of the original space was selected for expensive DFT labeling.
- High-Fidelity DFT Labeling: All selected structures were labeled with r2SCAN calculations using uniform computational settings, minimizing the functional and hardware-induced inconsistencies prevalent in prior datasets.
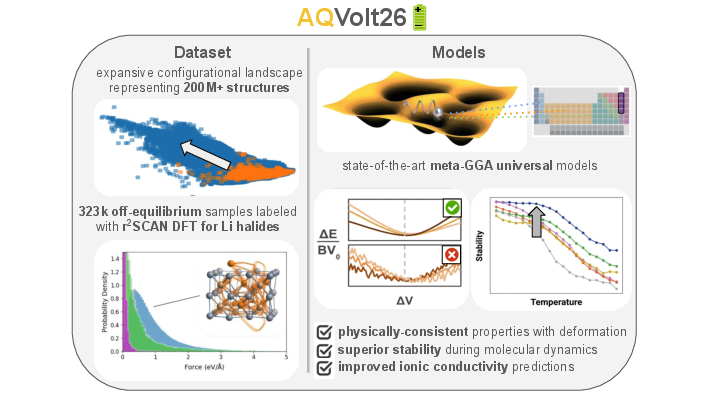
Figure 1: AQVolt26 encompasses 322,656 r2SCAN calculations for Li halides, co-trained with meta-GGA datasets, and demonstrates superior modeling of materials under strain, more stable MD at high temperature, and more accurate ionic conductivity relative to experiment.
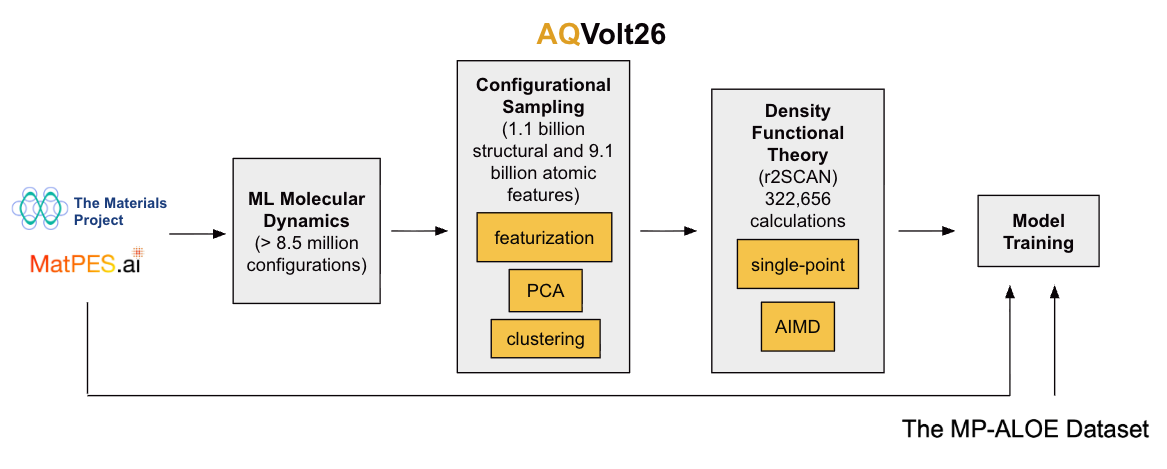
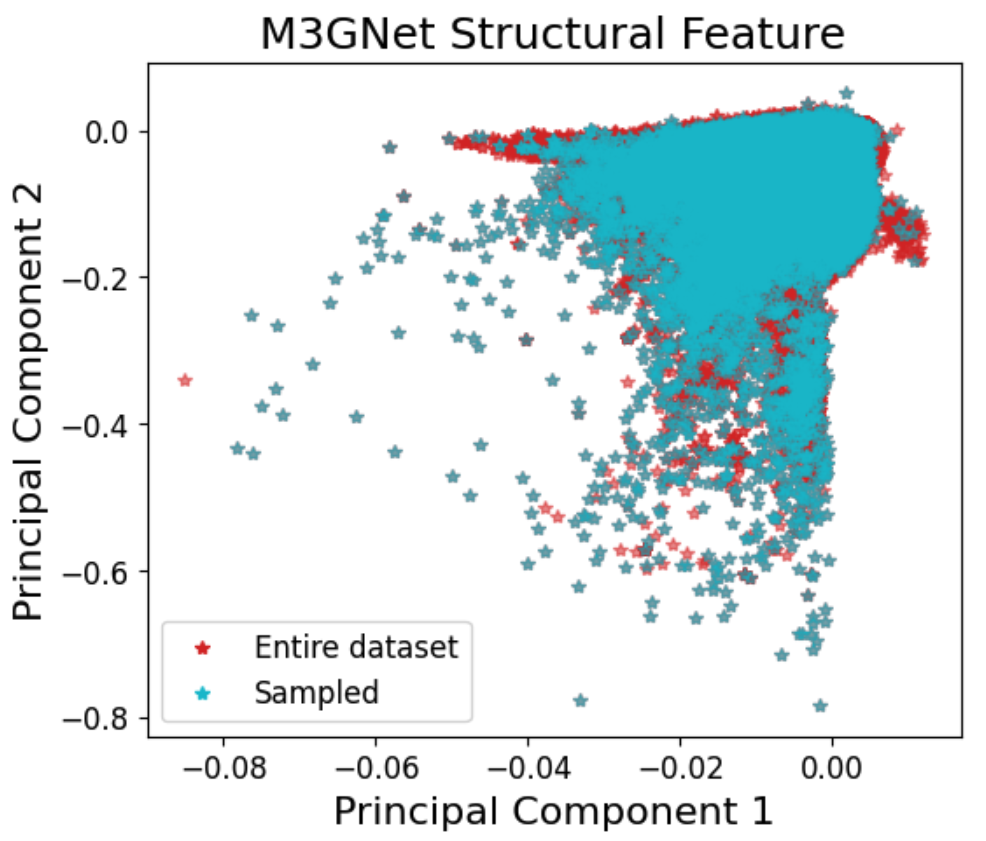
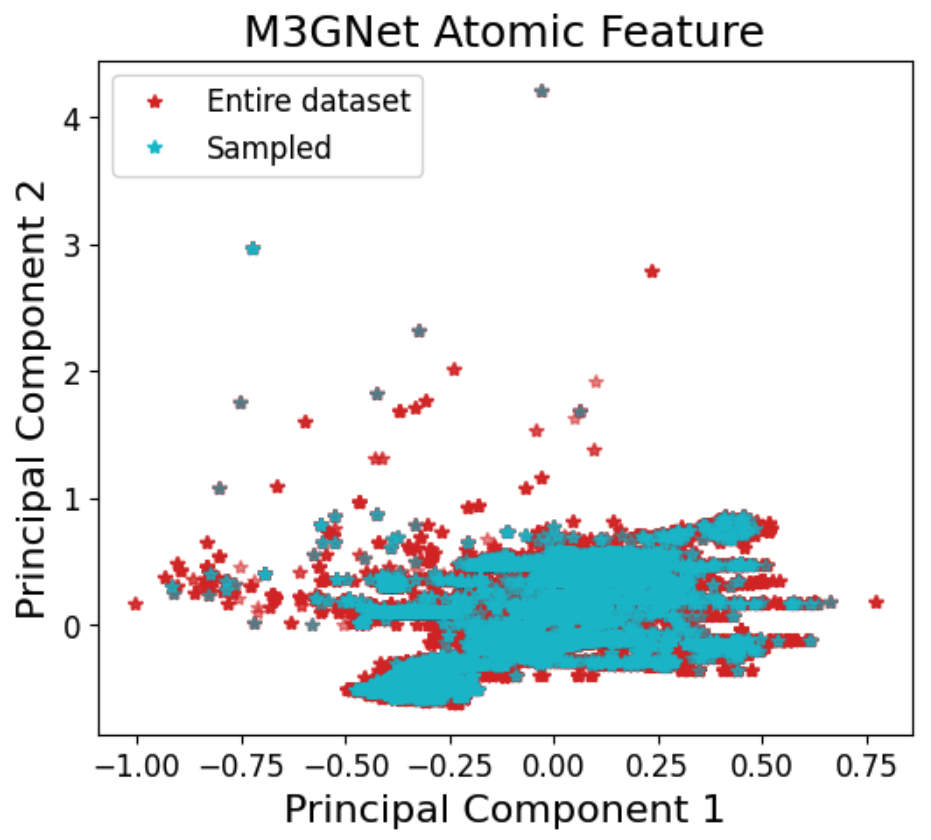
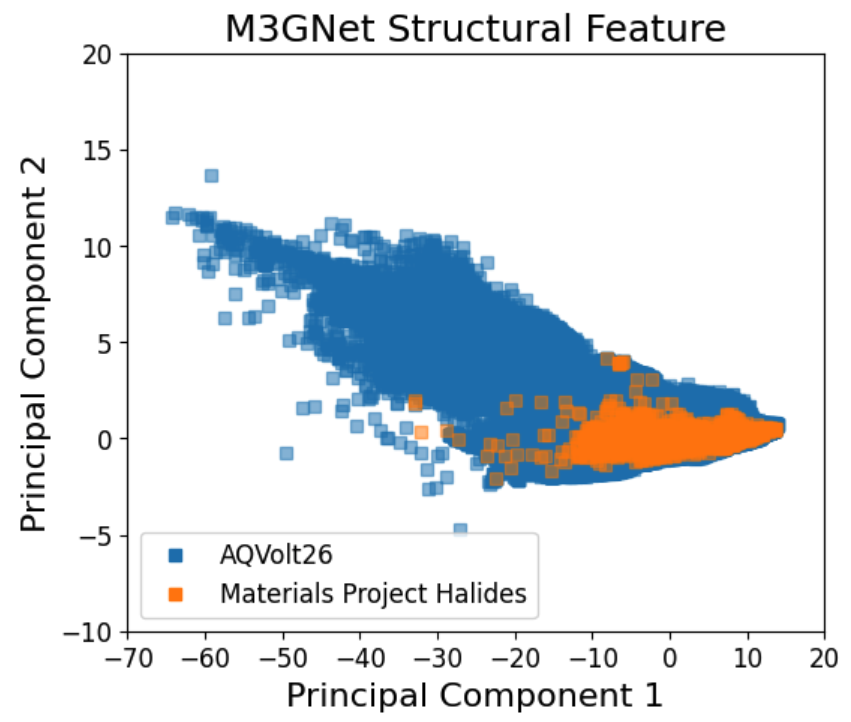
Figure 2: The AQVolt26 sampling and labeling pipeline visualized, highlighting surrogate-driven structural exploration and 2DIRECT-based dimensionality reduction for diverse, high-information data selection.
Compared to previous datasets (e.g., MatPES, MP-ALOE, and the Materials Project), AQVolt26 vastly expands the compositional and configurational coverage of Li halides, with a strong emphasis on multi-component, high-site-count materials and out-of-equilibrium structures.
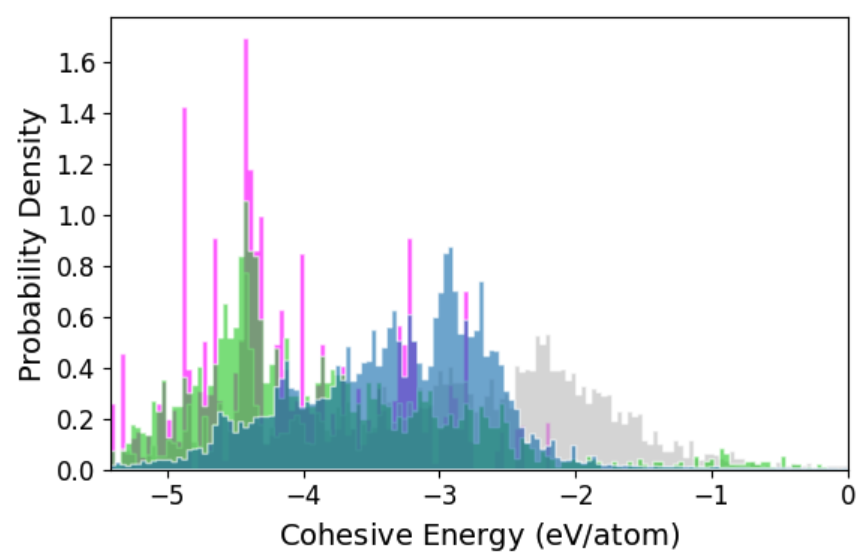
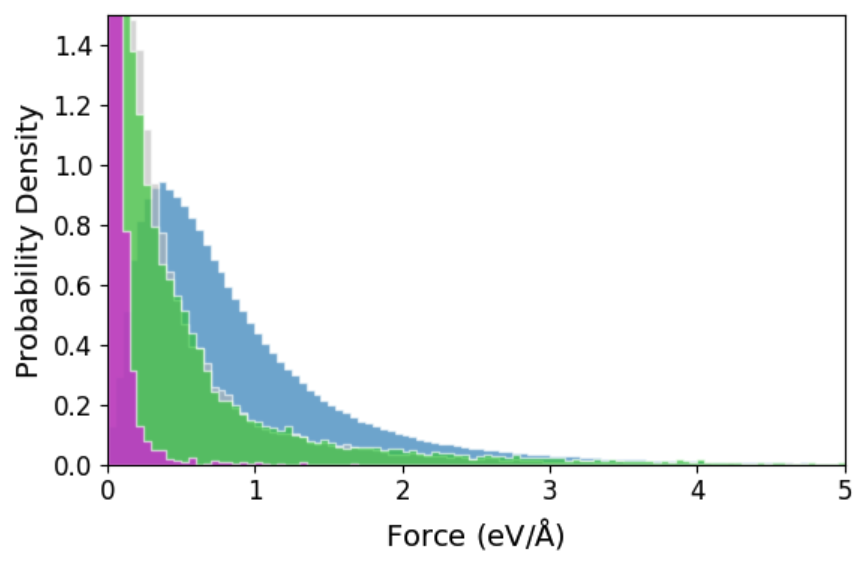
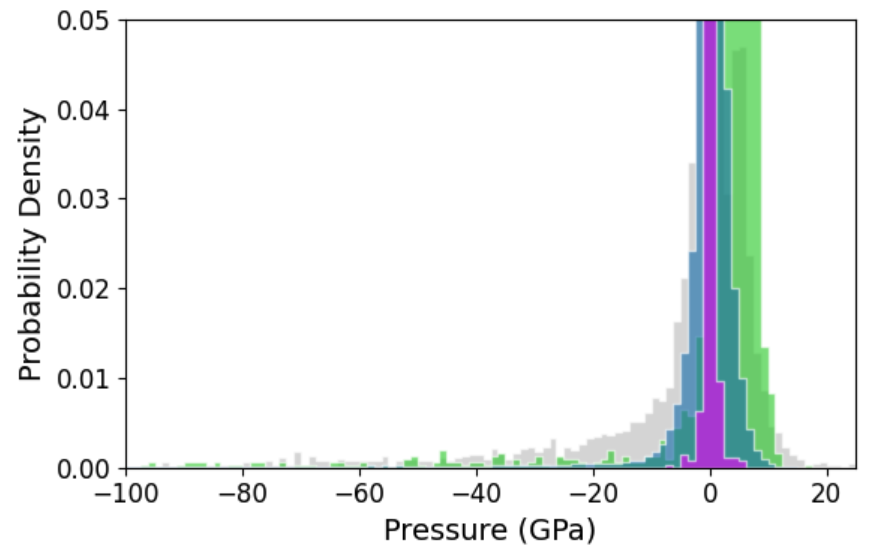

Figure 3: Cohesive energy, force, and pressure distributions for AQVolt26 (blue) broadening beyond other r2SCAN datasets toward high-temperature, off-equilibrium states vital for superionic transport exploration.
Model Training and Benchmarking
The study adopts the equivariant Smooth Energy Network (eSEN) architecture, leveraging a rigorously staged protocol (force pretraining followed by conservative fine-tuning). To maximize physical scope and reduce the risk of overfitting, eSEN models are co-trained with MatPES, MP-ALOE, and, optionally, Materials Project relaxations. The impact of dataset composition is systematically assessed by ablating MAP, MP-ALOE, and AQVolt26 in the training set.
- Benchmark Regimes: Models are benchmarked on multiple tasks—including off-equilibrium Li halides (AQVolt26), general off-equilibrium structures (MatPES/MP-ALOE), near-equilibrium relaxations (Materials Project, GNoME), and out-of-distribution high-temperature MD stability (GNoME).
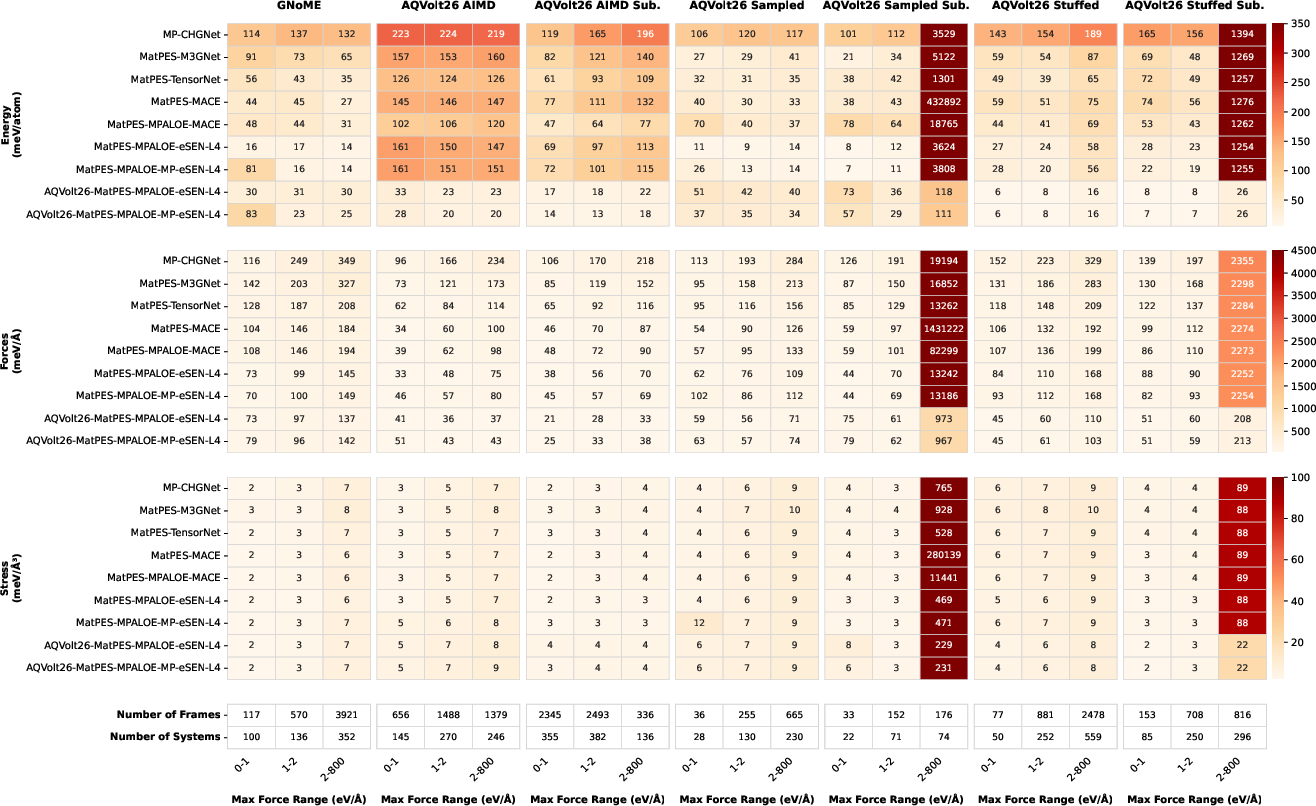
Figure 5: Energy, force, and stress prediction accuracy for various models, binned by DFT force magnitude and evaluated on diverse r2SCAN datasets, demonstrating the robust off-equilibrium performance of AQVolt26-eSEN models.
Notably, models trained only on near-equilibrium data (e.g., MP-CHGNet) rapidly deteriorate beyond low-force, stable configurations, while AQVolt26-trained eSEN architectures maintain lower errors in energy and force as perturbations increase.
Impact on High-Temperature MD Fidelity
A principal criterion for dataset utility is its ability to yield MLIPs that remain stable and predictive during high-temperature, long-timescale MD. The AQVolt26-eSEN models are systematically evaluated on controlled heating ramps (300–2,100 K). Across 25 Li halide and 100 halide-free GNoME structures, AQVolt26-eSEN exhibits high survival rates, evidencing robust dynamical guardrails even under extreme perturbation.
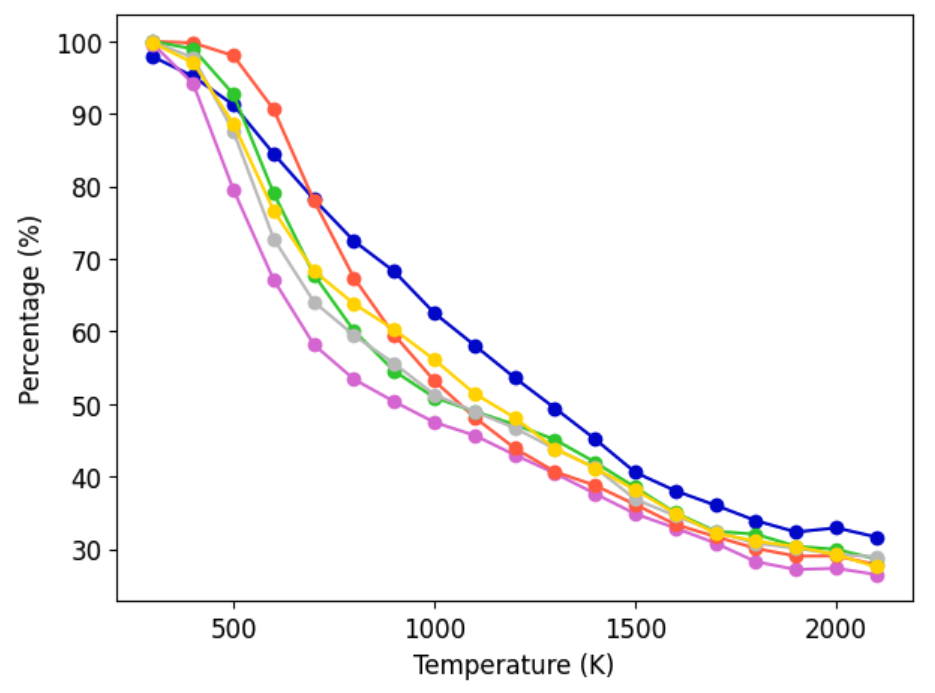
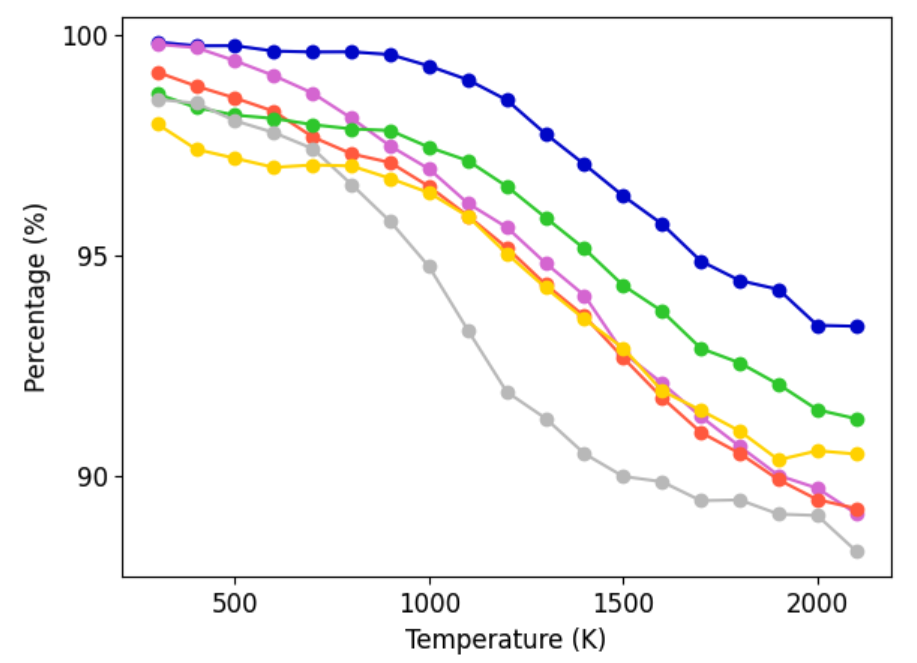

Figure 7: Survival curves for MLIPs during high-temperature MD ramps on Li halide and halide-free structures, with AQVolt26-eSEN maintaining superior or competitive stability across all temperatures.
Static lattice-strain benchmarks corroborate these findings: AQVolt26-eSEN displays near-zero failure under bulk hydrostatic deformations (±20%), affirming its physically consistent PES mapping.
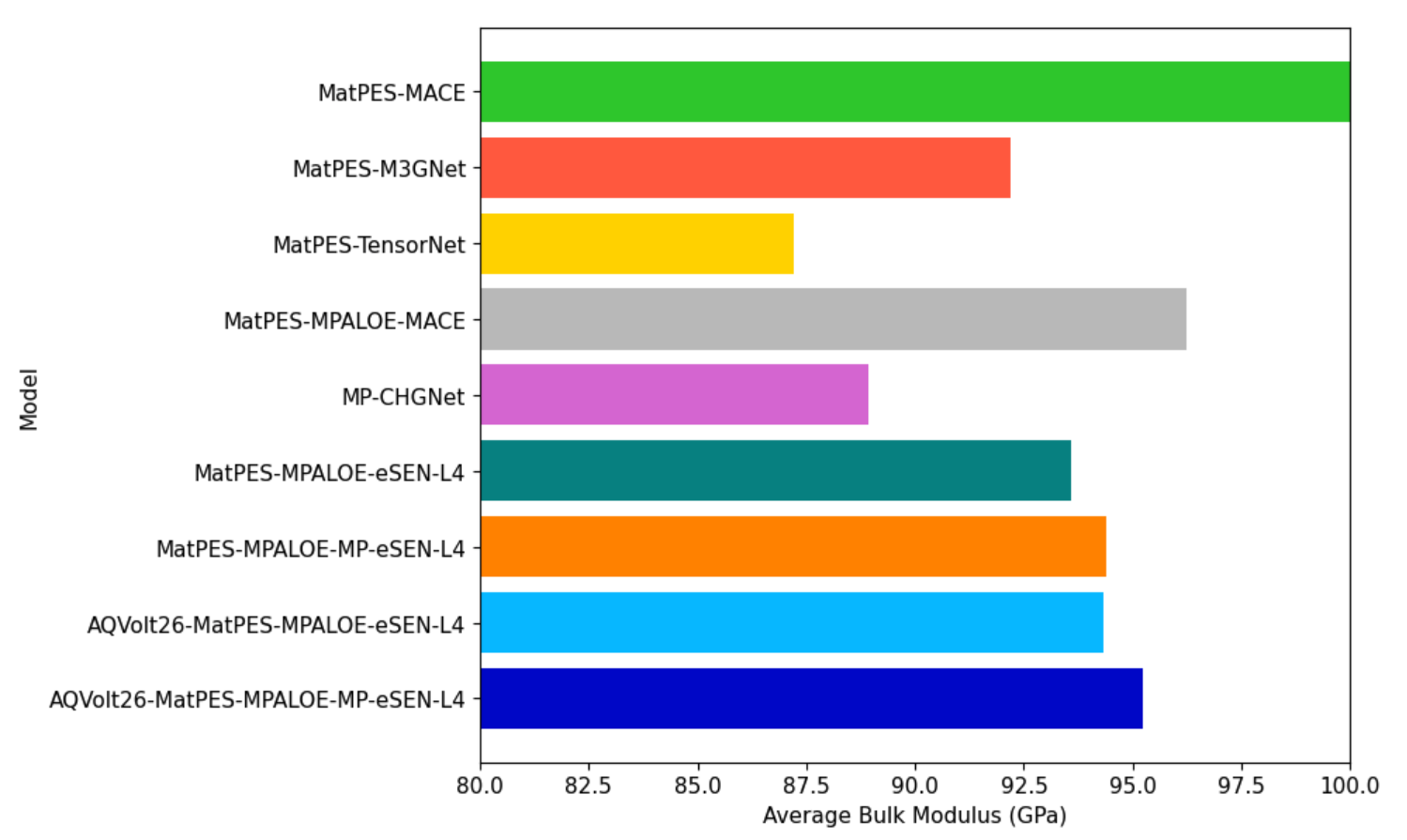
Figure 6: Bulk moduli evaluated from MLIP-Arena E–V scans indicating that off-equilibrium data induces stiffer response curves, capturing the steep PES features vital for resilience under perturbation.
Ground-State Precision vs. Dynamic Coverage
A longstanding concern is whether exposing MLIPs to highly strained, non-equilibrium structures might degrade their accuracy on equilibrium, ground-state properties (e.g., structure relaxations, energies). Structural fingerprints and energy prediction errors for GNoME-relaxed structures probe this effect. AQVolt26-eSEN models retain competitive geometric similarity and only a moderate increase in energetic error, with most predictions within acceptable margins for screening purposes.
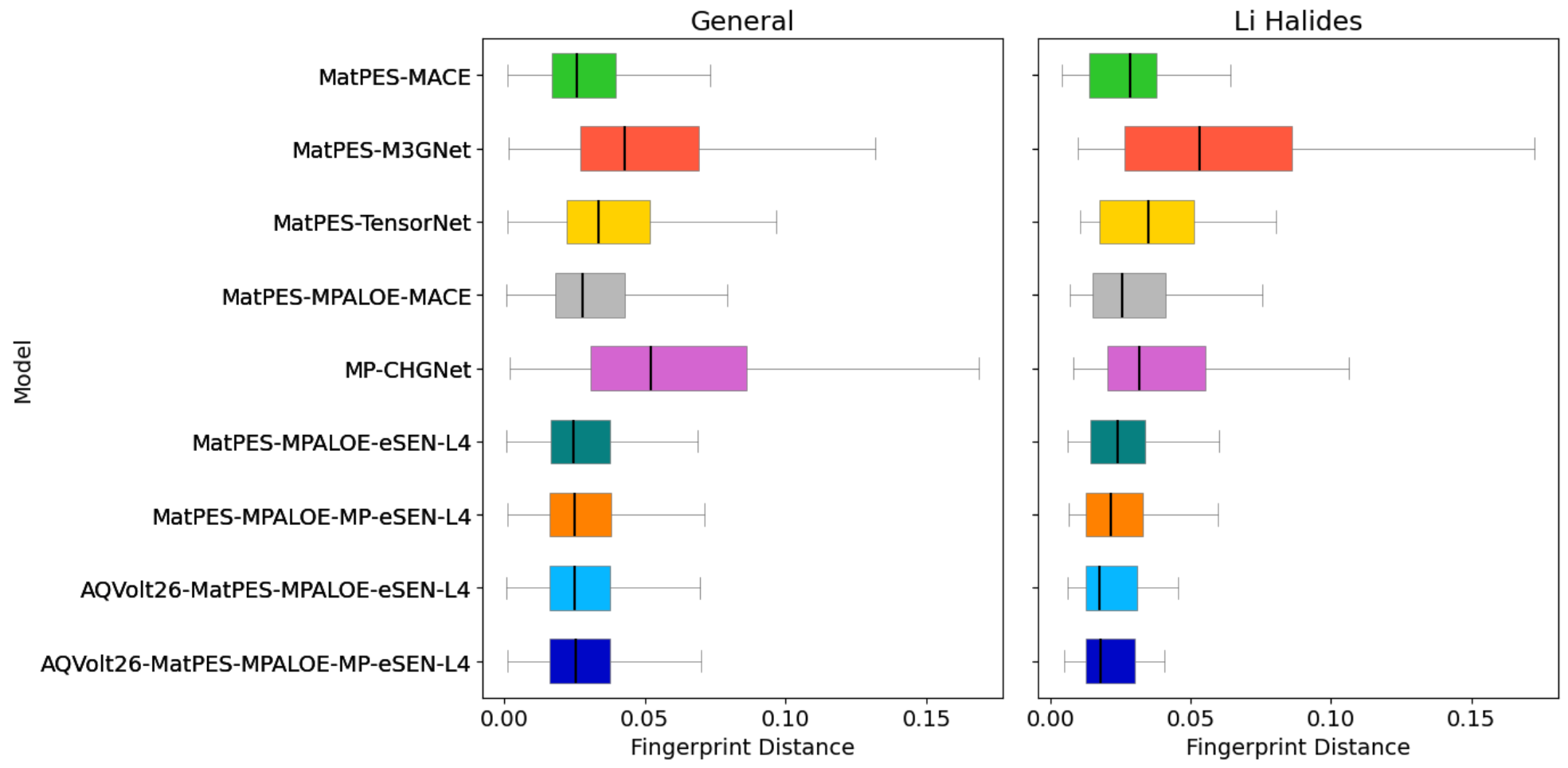
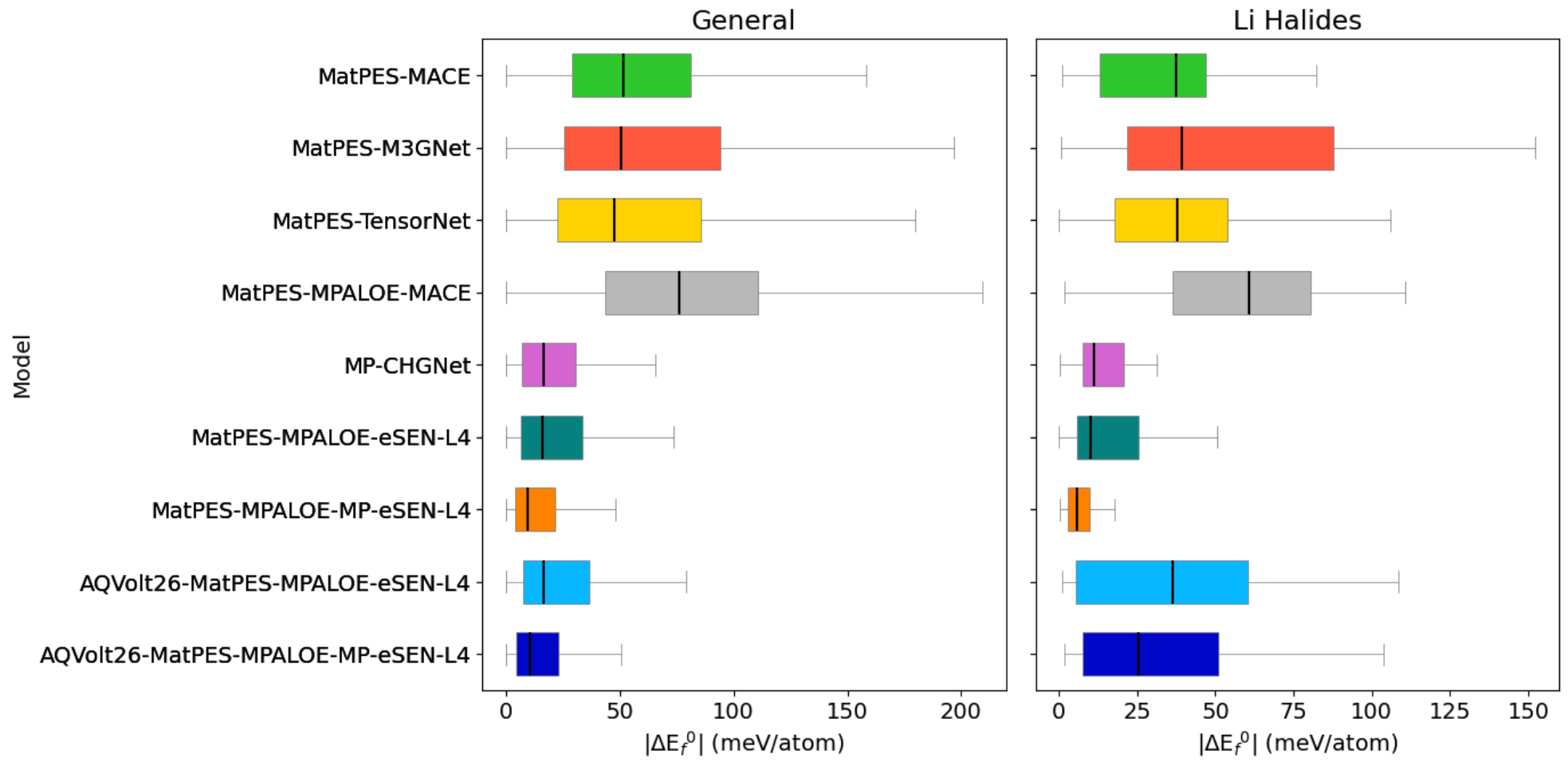
Figure 9: Fingerprint distances (top) and per-atom formation energy deviations (bottom) between MLIP-relaxed and DFT-relaxed GNoME structures, confirming the competitive ground-state quality retention in AQVolt26-trained models.
Downstream Materials Kinetics: Ionic Conductivity Prediction
An explicit motivation for AQVolt26 is improved quantitative prediction for properties depending on anharmonic, high-temperature dynamics—most notably Li+ ion conductivity. When evaluated against experimental OBELiX benchmarks, AQVolt26-eSEN achieves MAE of 0.6 mS/cm for Li halides versus 4.2 mS/cm for comparable MatPES-TensorNet potentials, with a substantial reduction in false positive conductivity predictions. For halogen-free chemistries, however, generalization is less pronounced, indicating potential trade-offs when extrapolating far from the training domain.
Theoretical Implications and Future Directions
The study bluntly demonstrates that dataset configurational and compositional coverage, rather than ML architecture, is the critical determinant of MLIP applicability across complex materials spaces. The universality of a potential can only be as broad as the data distribution supporting it, a principle echoed in recent foundational PES works ("A Foundational Potential Energy Surface Dataset for Materials" (Kaplan et al., 6 Mar 2025); "MP-ALOE: an r2SCAN dataset for universal machine learning interatomic potentials" (Kotsuka et al., 2023)).
AQVolt26 advances this paradigm by advocating for aggressive, domain-targeted sampling (e.g., high temperature, high perturbation for dynamically soft materials). This methodology is conceptually transferable to other superionic systems (e.g., sulfides, hydrides) where shallow PES and anharmonicity frustrate general-purpose MLIPs.
Conclusion
AQVolt26 delivers a comprehensive r2SCAN-level dataset and associated MLIP models tailored for the simulation of ionic transport and dynamic stability within lithium halide solid-state electrolytes. Through targeted off-equilibrium sampling and extensive DFT labeling, the resulting models deliver order-of-magnitude improvements on high-temperature Li halide tasks—without catastrophic compromise of equilibrium property prediction or stability on unrelated compositions. The work reifies the necessity of configurationally inclusive datasets for truly robust, application-ready MLIPs and lays a blueprint for domain-specific expansion of atomistic ML modeling in next-generation materials discovery.
References:
Kim et al., "AQVolt26: High-Temperature r2SCAN Halide Dataset for Universal ML Potentials and Solid-State Batteries" (2604.02524)
Kaplan et al., "A Foundational Potential Energy Surface Dataset for Materials" (Kaplan et al., 6 Mar 2025)
Kuner et al., "MP-ALOE: an r2SCAN dataset for universal machine learning interatomic potentials" (Kotsuka et al., 2023)