- The paper demonstrates that an LLM-driven agent can autonomously set up, execute, and refine all-atom MD simulations for polymer property prediction.
- It integrates RadonPy for molecular construction with GPU-powered LAMMPS via MCP, achieving validated predictions for T_g, density, and bulk modulus.
- The system exhibits dynamic error recovery and protocol adaptation, though challenges like cooling rate artifacts and kinetic trapping highlight areas for future improvement.
PolyJarvis: LLM-Driven Autonomy for Polymer Molecular Dynamics
System Architecture and Workflow Orchestration
PolyJarvis represents a significant methodological advance in the autonomous execution of all-atom MD simulations for polymer property prediction, uniquely integrating LLM reasoning with domain-specific simulation tools via the Model Context Protocol (MCP). The system architecture involves three principal components: the LLM agent (Claude Sonnet 4.5), a local MCP server wrapping RadonPy for molecular construction and force field handling, and a remote MCP server coordinating GPU-accelerated LAMMPS simulations on cloud infrastructure. This modular, client–server schema enables seamless bidirectional tool invocation and structured information transfer at every workflow stage, thus supporting fully autonomous polymer system setup, simulation, and property extraction from natural language inputs.
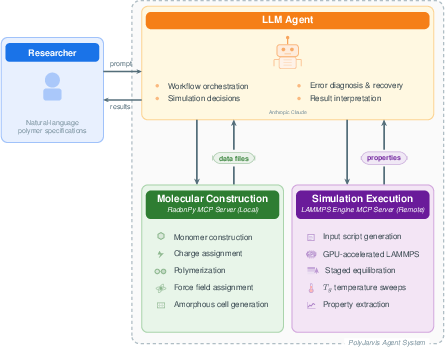
Figure 1: PolyJarvis system architecture, depicting LLM-mediated orchestration of RadonPy-based molecular construction and GPU-accelerated LAMMPS simulation via MCP servers.
Autonomous Simulation Protocols and Agent Decision-Making
The central innovation in PolyJarvis lies in LLM-driven, polymer-specific decision-making at each simulation stage. Upon receiving a natural language prompt specifying the target polymer—via name or SMILES—the agent classifies the system against PoLyInfo backbone categories, then autonomously determines the appropriate force field (GAFF2 or GAFF2_mod), charge assignment method (e.g., RESP or Gasteiger), and protocol parameters such as system size, density initialization, and electrostatics handling.
For each benchmark polymer (polyethylene [PE], atactic polystyrene [aPS], poly(methyl methacrylate) [PMMA], and poly(ethylene glycol) [PEG]), the agent adapts chain length (10 chains, n=62–150) and implements a staged equilibration protocol (NVT heating, NPT compression/decompression, annealing), refining these choices iteratively based on observed convergence and error diagnostics across replicate runs. Notably, PolyJarvis exhibits error recovery behaviors and protocol revision, such as correcting simulation instabilities, modifying annealing cycles, and resolving charge/spatial assignment failures without human intervention.

Figure 2: Example agent interaction, illustrating natural language progression from task specification to protocol clarification and automated reporting of property predictions with reference comparisons.
Quantitative Property Predictions and Experimental Benchmarking
Glass Transition Temperature (Tg)
Glass transition temperature predictions, derived via bilinear fitting of stepwise-cooled density–temperature profiles, were validated against reference values with stringent acceptance criteria (∣Tgsim−Tgexp∣≤20 K). PMMA exhibited close agreement (Tg=395 K, +10–18 K relative to experiment). aPS and PEG predictions consistently overestimated Tg by +43–+47 K (relative to upper experimental bounds), in line with known MD cooling-rate artifacts.
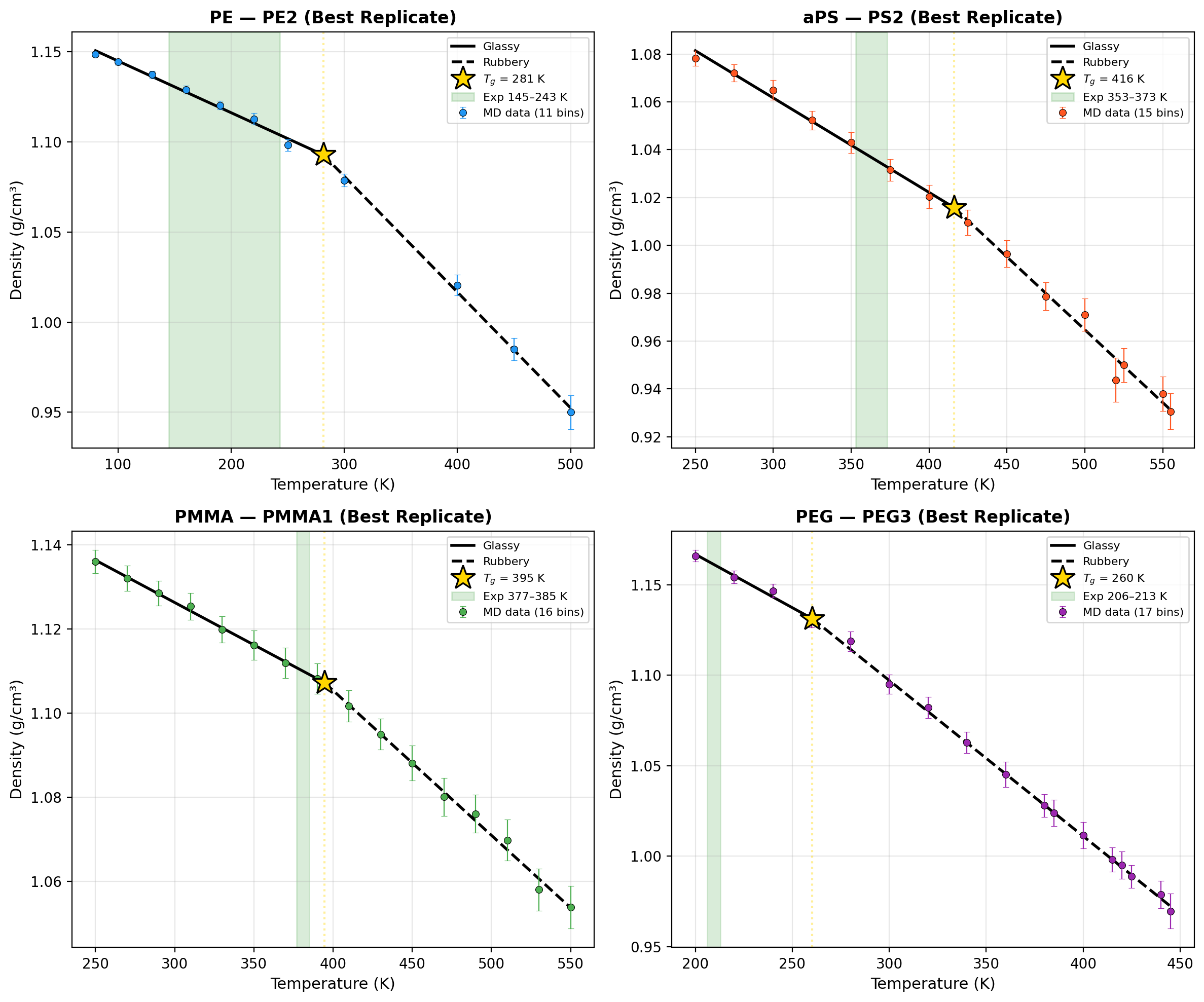
Figure 3: Density vs. temperature traces with bilinear fits and extracted Tg overlaid against literature experimental ranges.
Density
Density predictions at 300 K for aPS, PMMA, and PEG were within 0.1–4.8% of reference values, passing the predefined 5% error threshold; only PE showed significant overestimation (+25%), attributed to agent-induced kinetic trapping—a protocol adaptation failure.
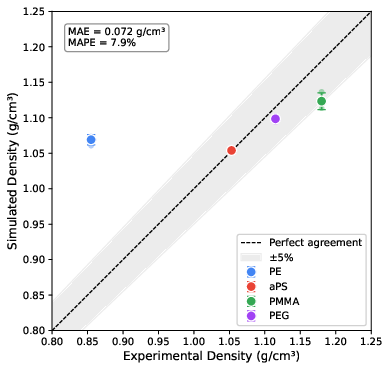
Figure 4: Parity plot comparing simulated and experimental density, ensemble means and run-level scatter, with acceptance band shaded.
Bulk Modulus
Bulk moduli, extracted from NPT trajectory volume fluctuations, showed agreement within 17–24% of experimental or well-benchmarked literature data for aPS, PMMA, and PEG (criterion: 30%). All predicted values for PE, despite its density overprediction, fell within the range of previous AA MD simulations.
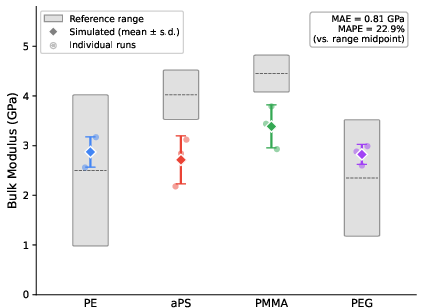
Figure 5: Predicted vs. reference bulk modulus at 300 K, ensemble means with error bars versus experimental bounds.
Structural Fidelity
All systems’ C–C radial distribution functions confirmed physically plausible amorphous packing and correct bond metrics, with convergence to g(r)=1 at long range and correct nearest-neighbor peak positions.
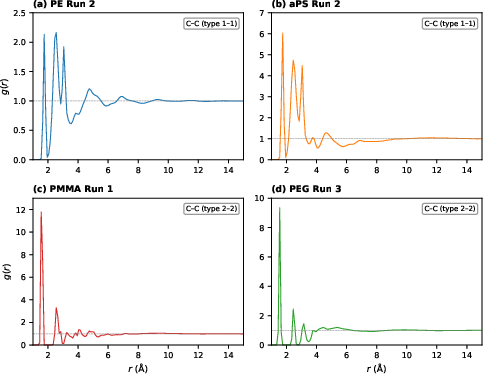
Figure 6: Carbon–carbon RDFs for best replicate of each polymer at 300 K, confirming absence of residual crystalline order.
Evaluation, Limitations, and Implications
Across eight property–polymer pairs benchmarked against directly comparable experiment, five met strict acceptance criteria. The remaining failures (PE density and aPS/PEG Tg) are attributed to persistent MD limitations (cooling-rate bias) and, in one case (PE), an agent calibration lapse leading to over-dense, kinetically trapped structures.
PolyJarvis provides robust evidence that LLM agents, with access to modular simulation toolchains and structured protocols (MCP), can autonomously execute non-trivial, chemically informed MD workflows. The agent’s ability to recover from common simulation failures, refine system parameters, and perform end-to-end property benchmarking underscores the practical viability of LLM-based scientific automation in computational polymer science.
The primary limitations are the restriction to several well-characterized amorphous homopolymers, protocol vulnerabilities (e.g., PE compaction), and LLM context/session constraints that may require sporadic human oversight. The impact of cooling-rate artifacts in Tg predictions remains an intrinsic MD issue, not specific to PolyJarvis’s agent design.
Outlook and Future Directions
PolyJarvis’s autonomous, LLM-driven approach opens avenues for integrating expert-level protocol reasoning with high-throughput, language-mediated computational materials design. Practical extensions include expansion to additional force fields (OPLS-AA, TraPPE-UA), formalization of adaptive protocol frameworks (potentially incorporating sequential decision processes), increased ensemble statistics, and validation for complex materials classes such as semicrystalline polymers and copolymers.
Theoretically, PolyJarvis demonstrates that LLM reasoning over structured tool APIs can substitute for human-in-the-loop domain expertise in multi-stage, stochastic simulation workflows. Future developments in agentic materials informatics may leverage model self-critique, extractable provenance, and integration with lab automation for closed-loop experimental design.
Conclusion
PolyJarvis establishes, with explicit benchmarking and quantitative validation, that LLM agents can autonomously orchestrate all-atom MD simulations yielding results consistent with best-in-class expert operation, within well-characterized domains. While system-level limitations persist, chiefly dynamical artifacts and protocol edge cases, the architectural paradigm of tool-augmented LLM decision-making marks a substantive advance in the automation of molecular simulation for polymer property prediction. Further developments will focus on enhancing adaptability, expanding chemical and force field coverage, and integrating with experimental data streams for truly autonomous, end-to-end materials discovery.