- The paper presents sparse autoencoders as tools for extracting interpretable latent features from complex single-cell gene expression data.
- It compares SAE architectures, showing Vanilla and ReLU perform robustly while TopK excels under specific hyperparameters.
- The findings emphasize the potential of SAEs in uncovering generative data structures and guiding biological interpretability.
Overview of Sparse Autoencoders in Gene Expression
Sparse autoencoders (SAEs) are explored in the context of extracting interpretable latent features from complex biological datasets, particularly single-cell gene expression data. The study explores the efficacy, limitations, and hyperparameter landscape of SAEs, examining their ability to extract generative variables from latent spaces and how they can uncover key biological signals.
Sparse Autoencoders and Application in Biological Data
SAEs have gained attention for their capacity to enhance interpretability by transforming dense embeddings into sparse, higher-dimensional representations, thereby disentangling learned features. This paper investigates the potential of SAEs for decomposing embeddings in highly complex biological domains, providing insights into the data structure and hidden variable recovery using simulated data inspired by sparse count data like single-cell gene expression.
The schematics of superpositions and SAEs (Figure 1) illustrate their function: converting samples featuring superposed features into disentangled representations in higher-dimensional spaces. Experiments reveal that variables closest to observable counts in the generative process were recovered most accurately. Recovery success is influenced by variable proximity and role within data generation.
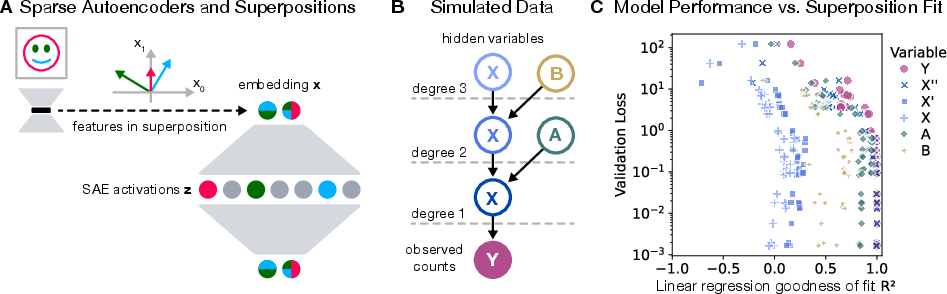
Figure 1: Sparse Autoencoders, data simulation, and hidden variable recovery.
Various SAE architectures, including Vanilla, ReLU, and TopK, underwent performance evaluations. Results indicated that Vanilla and ReLU configurations maintained robust reconstruction losses and activation performance across varying hyperparameters, while TopK models exhibited advantageous performance under specific conditions like lower learning rates and increased hidden dimensionality. Recovery efficiency and redundancy metrics showed disparities across architectures, highlighting Vanilla SAEs as achieving a favorable balance between recovery accuracy and redundancy minimization.
Implications for Discovering Data Variables and Structure
SAEs were applied to large simulation data to explore their recovery capacity for both data variables and generative structures. The connectivity matrix, derived from cosine similarities between SAE features and observables, was compared to established generative processes. Remarkably, SAE connectivity with observables offered substantial insight into the generative structure, even when individual generative variables were not fully recovered, showcasing SAE's potential in structural discovery in complex datasets.
Case Study on Single-cell Models: Scalable Interpretability
SAEs were tested on representations from pre-trained single-cell RNAseq and multi-omics models, yielding insights into biological processes through automatic annotation pipelines like scFeatureLens. These tools link SAE features with biological concepts to facilitate large-scale interpretability and hypothesis generation. Evaluations demonstrated that SAEs effectively extract biologically meaningful features critical for cellular processes, exemplified by manual and automated analyses.
The study stresses the practical value of SAEs in assessing and guiding cellular differentiation processes, validating feature importance via manual evaluations, and automated processes, enriching interpretation of gene sets within scRNAseq embeddings.
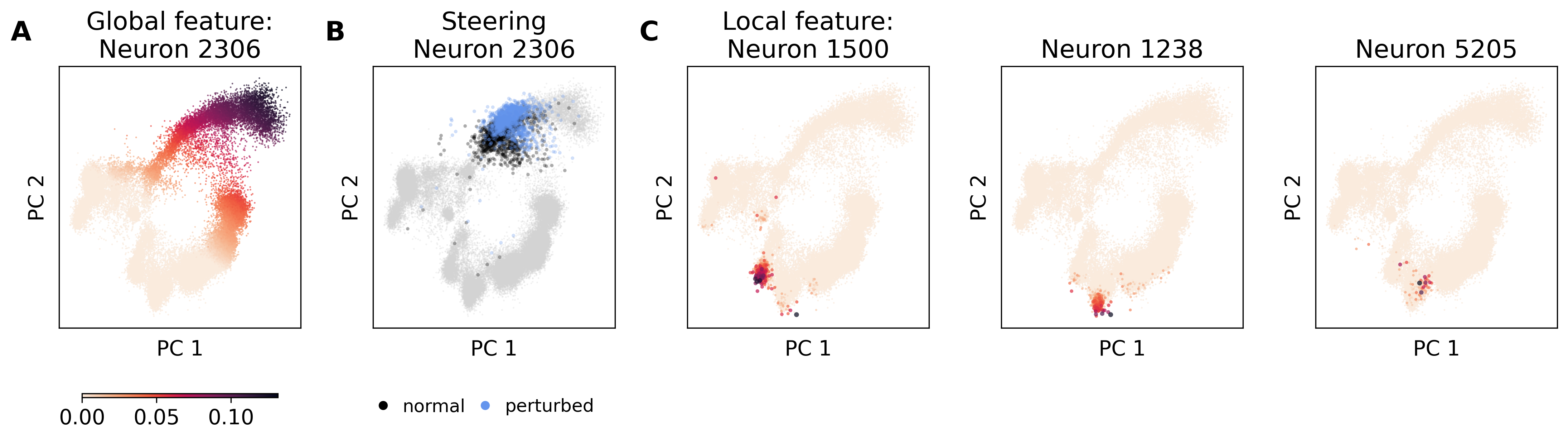
Figure 2: SAE features in multiDGD' bone marrow representation space.
Conclusion
Sparse autoencoders present valuable tools for interpreting complex biological representations, offering practical means for uncovering cellular processes and structures. While demonstrating effectiveness in separating observable features, their limitations in recovering deeper generative variables highlight challenges inherent in biological data decomposition. Nonetheless, their ability to organize latent spaces and facilitate biological understanding marks a noteworthy stride in biological interpretability research.
Future developments may target deeper understanding and evaluation of biological embedding structures, creating metrics for assessing biological meaningfulness, and exploring methods to circumvent barriers in latent variable recovery.
All code resources for reproducibility and the scFeatureLens tool are made available for the community, fostering advances in the interpretation of single-cell gene expression models.